1. Introduction
Rhodothermus is the type genus of the phylum Rhodothermaeota (Munoz et al., 2016). The aerobic bacterium R. marinus is thermophilic and moderately halophilic, growing optimally at 65 °C and 1–2% NaCl (Alfredsson et al., 1988). The bacterium has potential for biorefinery applications, as it possesses many features of importance for industrial bioconversion of recalcitrant 2nd and 3rd generation feedstock. This includes various biomass-degrading enzymes, as well as the capacity to produce interesting primary and secondary anabolic compounds, such as carotenoids (Ron et al., 2018) and exopolysaccharides (EPS) (Sardari et al., 2017). In addition, as a robust extremophile, R. marinus is adapted to growth at high temperatures which may be beneficial for biorefining of 2nd and 3rd generation feedstocks. High temperature increases the solubility of polysaccharides used as carbon sources and reduces the viscosity of fermentation broths. Consequently, it enables higher feedstock loads and facilitates enzymatic access to polysaccharides. In addition, growth at high temperatures in bioreactors mitigates scale-up problems of mixing and aeration, reduces costs of cooling, distillation and extraction and minimizes the danger of contamination of spoilage bacteria (López-Contreras et al., 2017).
To generate efficient biorefinery production strains, metabolic engineering is often required to modify the production profile of the microorganism. A few thermophiles have to date been the subject of metabolic engineering efforts, mainly anaerobic thermophiles for production of biofuels (and/or commodities), with encouraging results (Nordberg Karlsson et al., 2020). Tools for genetic engineering of the thermophilic aerobe R. marinus have been developed, and genes have both been heterologously expressed (Bjornsdottir et al., 2007) and deleted from the genome (Bjornsdottir et al., 2011).
Selection of a chassis species for metabolic engineering of pathways for production of compounds of industrial interest should be based on the metabolic characteristics of the organism, including both the substrate utilization range required for a particular feedstock, and the synthetic capabilities or potential. Anaerobic fermentative species are generally preferred for production of lower value commodity chemicals, such as simple organic acids and alcohols, which are typically waste products from catabolic metabolism. However, aerobes can carry a greater metabolic burden and are the organisms of choice for the heterotrophic production of complex secondary metabolites. This includes carotenoids, which are pigments, naturally produced by many plants, algae and bacteria. Currently, over 1100 carotenoid structures from more than 600 organisms are known (Yabuzaki, 2017). They are widely used as colorants and additives in the food, feed and cosmetic industries. Numerous studies have shown that carotenoids have potential health benefits, both in preventing and treating various diseases (Mein et al., 2008; Sathasivam and Ki, 2018). However, the biological functions of carotenoids are complex and other studies have reported conflicting results (Young and Lowe, 2018). Nevertheless, the global market for carotenoids has reached USD 1.5 billion and is predicted to continue growing (Markets And Markets, 2020).
R. marinus natively produces γ-carotenoids and their structures have been characterized (Lutnaes et al., 2004; Ron et al., 2018). Four variations of carotenoid glucoside esters were demonstrated, including that of salinixanthin. These native carotenoids are derived from γ-carotene, while most of the carotenoids of industrial interest are β-carotenoids. Astaxanthin, β-carotene, canthaxanthin, zeaxanthin, lycopene and lutein are in highest demand (Markets And Markets, 2020).
We previously carried out a bioinformatic analysis of the carotenoid biosynthetic pathway in R. marinus (Ron et al., 2018). The feasibility of modifying the pathway has also been demonstrated in a prior study, where the genes encoding the phytoene desaturase (CrtI) and phytoene synthase (CrtB) enzymes of the pathway were deleted, resulting in the colorless R. marinusstrain SB-71 (Bjornsdottir et al., 2011). The present study aims to clarify the function of the genes involved in the R. marinus carotenoid biosynthesis, and to engineer the pathway for lycopene production. Lycopene is one of the more demanded carotenoids on the market, used in cosmetics, pharmaceuticals and as a food coloring agent, and it is also the common precursor of γ- and β-carotenoids. For this purpose and to illustrate the potential of R. marinus as a biorefinery strain, lycopene production is an ideal first target for metabolic engineering.
2. Methods and materials
2.1. Strains, plasmids, media and culture conditions
The strains and plasmids used and generated in this study are listed in Table 1. For genetic modifications of R. marinus, strain SB-62 (ΔtrpBΔpurA) (Bjornsdottir et al., 2011) was used. Tryptophan selection was used in all cases, leaving the possibility of adenine complementation for further genetic modifications of the strains in the future. Genomic DNA from R. marinus ISCaR-493 and Thermus thermophilus HB8 as well as the plasmid vector pUC19 (Yanisch-Perron et al., 1985) were used for generating recombinant molecules. NEB Stable E. coli cells (New England BioLabs) were used for molecular cloning. The pRM3000 plasmid (Bjornsdottir et al., 2007) was used as a control in the R. marinus transformation experiments. All R. marinus cells were cultured at 65 °C and the liquid cultures set to shaking at 200 rpm (New Brunswick Innova 4400 Incubator Shaker). Medium 162 (Degryse et al., 1978) was used, with modifications (2 mM MgSO4and 0.2 mM CaCl2 in final volume) and additions of 1% NaCl and 0.053% NH4Cl. Two variations of the medium were used: A rich non-selective medium (R-medium) which contained 0.25% tryptone and 0.25% yeast extract and a selective agar medium (RS-medium), which contained 0.2% soluble starch, 0.2% casamino acids, vitamin solution (Degryse et al., 1978), 0.25% adenine and 2.5% agar. E. coli cells were cultured at 37 °C in LB-medium (Miller, 1972) with 100 μg/mL ampicillin.
Table 1. Stains and plasmids used in this study.
| Strain or plasmid | Details | Origin or reference |
|---|---|---|
| NEB Stable E. coli | Competent, High Efficiency | New England BioLabs |
| T. thermophilusHB8 | WT strain | Complete genome sequence available in GenBank (NC_006461.1) |
| R. marinusISCaR-493 | WT strain. Alternative names: PRI 493, MAT 493, DSM 16675 | Matís, (Bjornsdottir et al., 2005) |
| R. marinus SB-62 | Trp-Ade-, ISCaR-493 derivative (ΔtrpBΔpurA) | Matís, (Bjornsdottir et al., 2011) |
| R. marinus SB-71 | Trp+Ade-, colorless, SB-62 derivative(ΔtrpBΔpurAcrtBI’::trpB) | Matís, (Bjornsdottir et al., 2011) |
| R. marinus TK-1 | Trp+Ade-, SB-62 derivative (ΔtrpBΔpurAΔcrtYO::trpB) | This work |
| R. marinus TK-2 | Trp+Ade-, SB-62 derivative (ΔtrpBΔpurAΔcruF::trpB) | This work |
| R. marinus TK-3 | Trp+Ade-, SB-62 derivative (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) | This work |
| pUC19 | AmpR general cloning vector for E. coli | Yanisch-Perron et al. (1985) |
| pRM3000 | trpB+ R. marinus-E. coli shuttle vector | Matís, (Bjornsdottir et al., 2007) |
For the analysis of carotenoids, R. marinus strains TK-1, TK-2, TK-3 and SB-71 were grown on agar plates with modified medium 162 supplemented with 1% NaCl and 0.025 g/L adenine. After 48 h at 65 °C, the colonies were transferred to 5 mL liquid LB-medium, supplemented with 0.025% adenine, and cultivated at 65 °C for 24 h in 50 mL falcon tubes. The cells were then cultivated in baffled shake flasks at 200 rpm increasing the cultivation volume from 50 mL to 100 mL sequential cultivations, maintaining 10% (v/v) inoculation volume. The harvested cells were then split and transferred to two 500 mL bioreactors(Multifors 2, Infors) for cultivation with the following parameters: 65 °C, pH 7, 1 VVM aeration, 200 rpm stirrer rate cascaded with dissolved oxygen tension (DOT) that was set to 40%. The cultivation was terminated at the onset of the stationary phase, indicated by a sudden increase of DOT, at which the cell culture was set to cool at 10 °C. The cells were harvested by centrifugation at 4 °C at 10,000 g for 10 min, after which the pellets were frozen at -80 °C and lyophilized until constant weights were reached.
2.2. Design and generation of cloning molecules
Recombinant DNA molecules were designed using the plasmid vector pUC19 and amplified regions of the genome sequences of R. marinus ISCaR-493 (Matís, unpublished) and T. thermophilus HB8 (NC_006461.1). DNA was isolated from the strains using the MasterPure Complete DNA purification Kit (Lucigen). Primers (Table S1) were designed for the amplification of ~1500 bp 5′ and 3′ flanking regions of the genes targeted for deletion as well as the trpB gene (selection marker) and the crtB gene from T. thermophilus. The primers were designed to support HiFi DNA assembly (NEBuilder HiFi DNA Assembly Master Mix, New England BioLabs). The primers Gene5_5_F, Gene5_5_R (5′ flanking region), Gene1_3_F, Gene1_3_R (3′ flanking region), Gene5_Gene14_F and Gene5_Gene14_R (trpB gene) were used to amplify the fragments for the gene deletion cassette used to obtain strain TK-1 (ΔtrpBΔpurAΔcrtYO::trpB). The primers Gene9a_5_F, Gene9a_5_R (5′ flanking region), Gene9a_3_F, Gene9a_3_R (3′ flanking region), Gene9a_Gene14_F and Gene9a_Gene14_R (trpB gene) were used to amplify the fragments for the gene deletion cassette used to obtain strain TK-2 (ΔtrpBΔpurAΔcruF::trpB). The primers Gene9_5_F, Gene9_5_R (5′ flanking region), Gene9_3_F, Gene9_3_R (3’ flanking region), Gene9_Gene13_F, Gene9_Gene13_R (crtB gene from T. thermophilus), Gene9_Gene14_F and Gene9_Gene14_R (trpB gene) were used to amplify the fragments for the gene deletion/insertion cassette used to obtain strain TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus). All following enzymes, kits and cells in this section were obtained from New England BioLabs. Q5 High-Fidelity DNA polymerase was used in all amplifications according to the manufacturer’s instructions. The correct sizes of the resulting fragments were verified by electrophoresis and they were subsequently purified from gel using the Monarch DNA Gel Extraction Kit. The fragments were assembled into SmaI restricted pUC19 vector and introduced into competent E. coli cells (NEB stable) by chemical transformation. Positive clones were confirmed by verifying the presence of the trpB gene by PCR, using Taq DNA polymerase and the primers Gene14_Verify_F and Gene14_Verify_R (Table S1). Vectors were isolated from positive clones using the Monarch Plasmid Miniprep Kit. Linear inserts for R. marinus transformation for the construction of strains TK-1 and TK-2 were obtained by PCR, using the Q5 High-Fidelity DNA polymerase and the primers Gene5_5_F and Gene1_3_R, and TK2_Total_F and TK2_Total_R (Table S1), respectively. A linear insert for R. marinus transformation for the construction of strain TK-3 was obtained by digesting the plasmid with XbaI and KpnI-HF.
2.3. Transformation of R. marinus
R. marinus cells were prepared for electroporation as described elsewhere (Bjornsdottir et al., 2005). The electroporation protocol was followed, using the GenePulser Xcell electroporation system (Bio-Rad) with pulses delivered at 20 kV/cm 1 μg of DNA was used per transformation, in ≤5 μL and mixed carefully with 40 μL of washed cells. Negative (sterile MilliQ water) and positive (pRM3000) controls were included. Transformed cells were grown on selective agar plates (without tryptophan) for 3–5 days.
2.4. Verification of genotypes using PCR and sequencing
Positive R. marinus clones were verified by PCR, using Taq DNA polymerase. Modifications of strain TK-1 were verified using primers Gene7_Verify_F, Gene7_Verify_R, Gene5_Verify_F, Gene5_Verify_R, Gene4_Verify_F, Gene4_Verify_R, Gene14_Verify_F and Gene14_Verify_R (Table S1). Modifications in strains TK-2 and TK-3 were verified using the primers Gene9a_Verify_F, Gene9a_Verify_R, Gene9b_Verify_F, Gene9b_Verify_R, Gene13_Verify_F, Gene13_Verify_R, Gene14_Verify_F and Gene14_Verify_R (Table S1). Additionally, the modifications were confirmed by sequencing. Several PCR reactions were performed for strains SB-62 and TK-1 using the primers Gene1_Seq_F, Gene1_Seq_R, Gene4_Seq_F, Gene4_Seq_R, Gene5_Seq_F and Gene5_Seq_R (Table S1) and Q5 DNA polymerase. The resulting ampliconswere sequenced using the ABI3730 system (Applied Biosystems, Thermo Fisher Scientific). For strain TK-2, Q5 DNA polymerase and primers Gene9a_Seq_F and Gene9a_Seq_R (Table S1) were used to amplify a 5188 bp region. For strain TK-3, Q5 DNA polymerase and primers Gene9_Seq_F and Gene9_Seq_R (Table S1) were used to amplify a 4686 bp region. Sequencing libraries were made from the amplicons using the Nextera DNA (TK-2) and Nextera Flex (TK-3) methods (Illumina) and libraries were sequenced on the Illumina MiSeq sequencing platform using the V3 2 × 300 cycle chemistry. Obtained fragments were assembled using Geneious.
2.5. Carotenoid extraction
Carotenoids were extracted from R. marinus strains TK-1, TK-2, TK-3, SB-71 and ISCaR-493 using two methods. In the first method, aqueous cell suspensions were sonicated in an ice bath for 5 × 2 min, with 1-min rests in-between in order to keep cold sample conditions. After sonication the samples were mixed with ethyl acetate (1:1). In the second method, lyophilized cells were powdered with a glass rod before mixing with dichloromethane (25 mL solvent per g freeze-dried cells). All organic phase extracts were vacuum filtered before being dried by rotary evaporation (Heidolph instruments) at 80 rpm at 40 °C. The extracts were reconstituted in 3 mL of dichloromethane, filtered through a 0.2 μm PTFE syringe filter, flushed with N2 gas and stored at -80 °C until mass spectrometry analysis.
Carotenoids were additionally extracted from strains TK-1, TK-2, TK-3, SB-71 and ISCaR-493, for absorbance spectra analysis, by mixing full loops of cells from agar plates with hexane:acetone (1:1) and sonicating in a bath for 20 min. This was done in triplicates.
2.6. Carotenoid analysis
The carotenoid extracts were analyzed by means of supercritical fluid chromatography – mass spectrometry (MS) in an Ultra Performance Convergence Chromatography system (UPC2) coupled to a quadrupole – orthogonal acceleration time-of-flight tandem mass spectrometer (XEVO-G2 Q-TOF) with an electrospray ion source, both instruments from Waters (Mildford, MA, USA). Both systems were controlled, and all data was analyzed, with MassLynx™ (v 4.1, SCN 77; Waters). The chromatographic method used was based on a modification of the method described in (Jumaah et al., 2016). Briefly, the chromatographic separation was achieved using Acquity UPC2 Torus 1-Aminoanthracene (100 mm × 3 mm, 1.7 μm) column from Waters. The mobile phase consisted of (A) CO2 and (B) methanol in a gradient elution analysis programmed as follows: 0–0.5 min, 5% (B); 0.5–2.5 min, 5–15% (B); 2.5–8 min, 15% (B); 8–9 min, 15-5% (B); and 9–10 min, 5% (B). The flow rate was 1.5 mL min-1, column temperature was kept at 56 °C and backpressure at 160 bar. Ammonium formate (10 mM) in methanol was used as a make-up solvent at a flowrate of 0.5 mL min-1. The total run-time of the program was 10 min.
UV-Vis spectra were recorded in the range of 200–500 nm by the diode array detector (DAD). The electrospray ionization (ESI) ion source was operated in positive mode and full-scan MS spectra were obtained by scanning the range m/z 50–1000. The mass spectrometer was calibrated with sodium formate. Centroid mode data was collected after mass correction during acquisition using an external reference comprising of 10 μL/min solution of leucine-enkephalin (2 ng/μL). The capillary and cone voltage were set at 3 kV and 40 V, respectively. Nitrogen was used as both cone gas (50 L/h) and desolvation gas (800 L/h). The source and desolvation temperature were set at 150 and 300 °C, respectively. Simultaneous acquisition of exact mass at high and low collision energy, MSE (where E represents collision energy), was used to obtain full scan accurate mass fragment, precursor ion, and neutral loss information. The collision energy in function 1 (low energy) was off while in function 2 (high energy) and the collision energy ranged between 15 and 60 V. MS/MS analysis of the carotenoids was performed with the quadropole set at m/z 536 in MSEmode.
The absorbance spectra (350–600 nm) of the carotenoids of strains TK-1, TK-2, TK-3, SB-71 and ISCaR-493, extracted with hexane:acetone (1:1), were analyzed in a benchtop spectrophotometer (MULTISKAN Sky, Thermo Scientific), using 1 cm cuvette. This was done to investigate the profile of the spectra and Amaxvalues of obtained peaks.
Lyophilized TK-3 cells from shake flask cultivations were weighed before organic solvent extraction and reconstitution in chloroform. The extracts were then analyzed by a spectrophotometer at 485 nm. The lycopene concentration was calculated using Beer’s law with the molar attenuation coefficient of 150855 L mol-1 cm-1 for lycopene in chloroform (Naviglio et al., 2008). Lycopene, α-, β- and γ-carotene standards were of analytical grade quality (Supelco).
3. Results
3.1. Bioinformatic analysis of genes encoding carotenoid biosynthetic enzymes
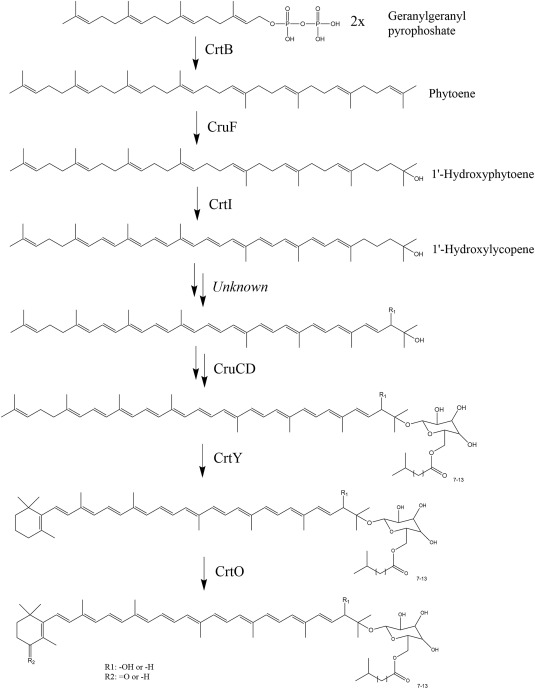
Several strains of the thermophilic bacterium R. marinus produce carotenoids derived from γ-carotenoid. Previous gene homology searches for known carotenoid biosynthetic genes using the genomic data of the type strain R. marinus DSM 4252T, resulted in the discovery of genes encoding homologues to enzymes in the carotenoid biosynthesis pathways from other species. This allowed the identification of a gene cluster, with two adjacent operons, one small and one larger (Bjornsdottir et al., 2011; Ron et al., 2018). In this work, the bioinformatic analysis of genomic sequence data was expanded, enabling further identification of the enzymes in the biosynthetic pathway for carotenoid production in R. marinus (Fig. 1).
 Fig. 1. Proposed carotenoid biosynthetic pathway in R. marinus, based on (Ron et al., 2018), with added information obtained in this study. The molecular structures and abbreviated enzyme names are shown, phytoene synthase (CrtB), 1’,2’-hydratase (CruF), phytoene desaturase (CrtI), glycosyltransferase(CruC), acyltransferase (CruD), lycopene cyclase (CrtY) and carotene ketolase (CrtO).
Fig. 1. Proposed carotenoid biosynthetic pathway in R. marinus, based on (Ron et al., 2018), with added information obtained in this study. The molecular structures and abbreviated enzyme names are shown, phytoene synthase (CrtB), 1’,2’-hydratase (CruF), phytoene desaturase (CrtI), glycosyltransferase(CruC), acyltransferase (CruD), lycopene cyclase (CrtY) and carotene ketolase (CrtO).Here, we report the identification of the cruF gene in the gene cluster (Fig. 2), which likely acts as a 1′,2′-hydratase (Sun et al., 2009). This gene was not identified in the previous work because it is fused together with the phytoene synthase (crtB) gene (Fig. 2A, gene 9) and together they were annotated as a phytoene synthase. These genes are usually separate, while cruF in R. marinus is without a stop codon and fused with the downstream crtB in one open reading frame (ORF). The fused genes, along with the carotenoid genes acyltransferase(cruD) and phytoene desaturase (crtI) (Fig. 2A, genes 9, 11 and 12, respectively) are located in the smaller operon in the gene cluster. Two additional genes are also present in the smaller operon, encoding products showing homology to hypothetical proteins (gene 10) and a MerR family transcriptional regulator from T. thermophilus (31% similarity) (gene 8).
 Fig. 2. The carotenoid gene cluster in the R. marinus stain SB-62 (ΔtrpBΔpurA) (A) and three different genetically modified strains: mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) (B), mutant TK-2 (ΔtrpBΔpurAΔcruF::trpB) (C) and mutant TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) (D). Genes involved in the carotenoid biosynthesis are colored grey. Gene deletions and insertions were verified using PCR (1–14) and the sequencing of PCR products (a–g). Pictures showing gel electrophoresis following PCR are shown to the right side of the gene clusters. Lane M1: 100 bp DNA ladder. Lane M2: 1 kb DNA ladder. Unedited electrophoresis pictures and sequencing results are shown in the supplementary file (Figs. S1–S11).
Fig. 2. The carotenoid gene cluster in the R. marinus stain SB-62 (ΔtrpBΔpurA) (A) and three different genetically modified strains: mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) (B), mutant TK-2 (ΔtrpBΔpurAΔcruF::trpB) (C) and mutant TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) (D). Genes involved in the carotenoid biosynthesis are colored grey. Gene deletions and insertions were verified using PCR (1–14) and the sequencing of PCR products (a–g). Pictures showing gel electrophoresis following PCR are shown to the right side of the gene clusters. Lane M1: 100 bp DNA ladder. Lane M2: 1 kb DNA ladder. Unedited electrophoresis pictures and sequencing results are shown in the supplementary file (Figs. S1–S11).Two genes in the larger operon were also identified based on sequence homology, with high sequence similarity to the known carotenoid genes encoding lycopene cyclase (crtY) (50%) (Fig. 2A, gene 2) and carotene ketolase (crtO) (60%) (Fig. 2A, gene 3). Five other genes (Fig. 2A, genes 1, 4–7) were found in this larger operon, but since they lack sequence homology to known genes in carotenoid biosynthetic pathways, their role in the pathway remains unknown. They were annotated as hypothetical protein (gene 1), NAD dependent epimerase (gene 4), FAD dependent oxidoreductase (gene 5), which showed slight homology (36% similarity) to crtI, deoxyribodipyrimidine photo-lyase (gene 6) and short-chain dehydrogenase/reductase (gene 7). Several known carotenoid biosynthetic genes belong to the crtI gene family, including crtO(gene 3, above) and also 3′,4′-desaturase, an enzyme for which the corresponding gene is not identified in R. marinus. The native structures of the carotenoids in R. marinus suggest that a 3′,4′-desaturase is a part of the biosynthetic pathway, but since the crtI homology of gene 5 is low and there is no other evidence so far that it encodes a 3′,4′-desaturase, this step in the pathway is still labeled as unknown (Fig. 1).
3.2. Genetic modifications to alter the carotenoid production
Three different genetic modifications were performed in the carotenoid gene cluster of R. marinus strain SB-62 (ΔtrpBΔpurA) (Fig. 2). The target genes were deleted and replaced with the selective marker trpB (encoding the tryptophan synthase beta chain), using double crossover homologous recombination using linear insertion cassettes. In the first modification, a 5890 bp region was deleted from the larger operon, resulting in the R. marinus mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) (Fig. 2B). This region includes the genes crtY (gene 2) and crtO (gene 3) which encode the enzymes responsible for the 4-keto β-ionone ring modifications displayed on the left side of the carotene backbone (Fig. 1), as well as gene 5, where the deduced amino acid sequence is showing slight homology to crtI (see section 3.1). The other two modifications involved knocking out the cruF gene (1′,2′-hydratase), which encodes one of the enzymes that modify the right side of the carotene backbone (Fig. 1). In the first cruF modification, only the cruF part of the gene was deleted, leaving the crtBpart intact, resulting in the R. marinus mutant TK-2 (ΔtrpBΔpurAΔcruF::trpB) (Fig. 2C). The crtB part of the gene includes a start codon, which raises the question if it could encode a functional enzyme without the cruF part. In the second cruF modification, both the cruF and crtB parts of the gene were deleted and the crtB gene from Thermus thermophilus strain HB8 was inserted, resulting in the R. marinus mutant TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) (Fig. 2D). This modification was designed in case the native CrtB enzyme in TK-2 would be non- or low functioning after the deletion of the cruF part.
Successful genetic modifications were verified by amplifying target regions from the genomes of putative mutants, and sequencing. PCR reactions amplifying several genes in the cluster (genes 1, 4, 5, 7, 9a (cruF), 9b (crtB), 13 and 14 in Fig. 2), using primers designed to bind inside each gene, were performed for all three mutants and strain SB-62. For strain SB-62 all reactions resulted in the expected sized amplicons, except for the crtB gene from T. thermophilus and the trpB gene (Fig. 2A). The presence of an amplicon using primers for amplifying the crtB gene from T. thermophilus in R. marinus was not expected. However, in all R. marinus strains the same sized unknown amplicon (~300 bp) was obtained and was subsequently sequenced. This showed that the amplification occurred from a location on the chromosome distant from the carotenoid gene cluster (Fig. S9). This gene did not show homology to crtB from T. thermophilus. The primers used to amplify the crtB gene from T. thermophilusdid however show homology to this region, which explains the unexpected amplification. The trpB gene was not amplified in strain SB-62 which was to be expected since it has the trpB gene deleted, enabling us to use it as a selective marker.
For strain TK-1 (ΔtrpBΔpurAΔcrtYO::trpB), the PCR results showed two main differences compared to strain SB-62 (Fig. 2B). First, genes 1, 4 and 5 were not amplified, which indicated the successful deletion of the region. Second, the reaction for the trpB gene (gene 14) did result in the expected sized amplicon, indicating the insertion of the selection marker. To further verify this, additional PCR reactions (a – f in Fig. 2) were performed for strains SB-62 and TK-1 and the products were sequenced (Figs. S5 and S6).
For strains TK-2 (ΔtrpBΔpurAΔcruF::trpB) and TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus), the cruF part of gene 9 was not amplified, while gene 14 was, indicating the expected substitution of cruF for trpB. Additionally, an amplicon of the expected size for gene 13 (crtB from T. thermophilus) was observed in strain TK-3. An identical amplicon was observed from T. thermophilus (Fig. S1). The native crtB gene (second half of gene 9) was deleted from strain TK-3 and the amplicon seen in strain SB-62 (~600 bp) was not amplified in strain TK-3. However, a smaller unknown amplicon (~400 bp) was obtained and was subsequently sequenced. The results showed that the amplification did not occur from the carotenoid gene cluster (Fig. S8), but in a gene on the chromosome distant from the cluster. This gene did not show homology to the crtB gene, but the primers used to amplify crtB in R. marinus did show homology to this region, which explains the unexpected amplification. To further verify the successful modifications in strains TK-2 and TK-3, additional PCR reactions were performed (g in Fig. 2) and the resulting products were sequenced (Figs. S10 and S11).
3.3. Analysis of carotenoids by UHPSFC-DAD-QTOF/MS and spectrophotometry
Absorbance spectra of carotenoids from strains TK-1 (ΔtrpBΔpurAΔcrtYO::trpB), TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) and ISCaR-493 extracted with hexane:acetone (1:1), showed Amax for strain ISCaR-493 at 478 nm, for TK-1 at 488 nm and for TK-3 at 472 nm, with an additional peak at 502 nm (Fig. 3 and Figs. S17-S19). The spectra for TK-3 is identical to previously published spectra for lycopene (Britton et al., 2004). Absorbance between 350 and 600 nm was neither observed for strain TK-2 (ΔtrpBΔpurAΔcruF::trpB) nor SB-71 (ΔtrpBΔpurAcrtBI’::trpB).
 Fig. 3. Carotenoid structures identified in R. marinus strain ISCaR-493 (A), mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) (B) and mutant TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) (C). The top row shows chemical formulas of identified carotenoids, corresponding theoretical masses and detected masses by MS. The middle row shows absorbance spectra from 350 to 600 nm of carotenoids isolated with acetone:hexane (1:1) and Amax values of obtained peaks. Graphs with both axes are shown in Figs. S17-S19. The bottom row shows the chemical structures of identified carotenoids: Native carotenoids (4-keto 2‘-hydroxy β,ψ-carotene acyl glycoside), modified lycopene (2’-hydroxy ψ,ψ-carotene acyl glycoside) and lycopene. For more information on R1 and R2, refer to Fig. 1.
Fig. 3. Carotenoid structures identified in R. marinus strain ISCaR-493 (A), mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) (B) and mutant TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) (C). The top row shows chemical formulas of identified carotenoids, corresponding theoretical masses and detected masses by MS. The middle row shows absorbance spectra from 350 to 600 nm of carotenoids isolated with acetone:hexane (1:1) and Amax values of obtained peaks. Graphs with both axes are shown in Figs. S17-S19. The bottom row shows the chemical structures of identified carotenoids: Native carotenoids (4-keto 2‘-hydroxy β,ψ-carotene acyl glycoside), modified lycopene (2’-hydroxy ψ,ψ-carotene acyl glycoside) and lycopene. For more information on R1 and R2, refer to Fig. 1.Extracts from R. marinus strains TK-1, TK-2, TK-3, SB-71 and ISCaR-493 were analyzed by UHPSFC-DAD-QTOF/MS. By DAD, it was not possible to detect any peaks between 400 and 600 nm for strains TK-2 and SB-71. Moreover, the mass spectrometer did not detect any masses corresponding to the native carotenoids nor masses corresponding to common C40 carotenoids. It was therefore concluded that TK-2 (ΔtrpBΔpurAΔcruF::trpB), like SB-71 (ΔtrpBΔpurAcrtBI’::trpB), does not produce carotenoids or produces them in very low concentrations. This was expected for SB-71 since the genes crtB and crtI (Fig. 1) were knocked-out (Bjornsdottir et al., 2011). For TK-2, these results indicated that the crtB part of the cruF-crtB gene (Fig. 2, gene 9) does not encode an active enzyme without the cruF part.
Two of the native carotenoids were detected in the TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) extract. The first detected carotenoid (tR 2.48) had a mass (m/z 910.672, Δ3.2 mDa, Δ3.5 ppm) matching the β,ψ-carotenoid acyl glucoside without the 2′-hydroxyl group previously identified in R. marinus.MS/MS fragmentation confirmed this by detection of the carotenoid fragment (C40H55) but not the 4-keto β-ionone ring fragment (m/z 203.1436) found in salinixanthin (Ron et al., 2018). Moreover, the characteristic m/z 28 pattern, resulting from acyl group length variation, was detected for C11 (m/z 882.637, Δ2.4 mDa), C13 (m/z 910.672, Δ3.2 mDa), C15 (m/z 938.703, Δ2.9 mDa), and C17 (m/z 966.732, Δ0.8 mDa). The other detected carotenoid (tR 2.83 min) in the TK-1 extract had a mass identical to that of the native 2′-hydroxyl version of the above mentioned β,ψ-carotenoid acyl glucoside (m/z 926.660 Δ4.1 mDa). This carotenoid followed a similar pattern of acyl length loss and MS/MS fragmentation as the native non-hydroxylated β,ψ-carotenoid acyl glucoside identified in a previous study (Ron et al., 2018). Salinixanthin in both the 2′-hydroxyl and non-hydroxylated forms was detected in TK-1. These results suggest that all the native modifications displayed on the right side of the carotene backbone in Fig. 1, including the C-3′,4′ double bond, are still present in TK-1. This refutes the hypothesis that gene 5 encodes a 3′,4′-desaturase (see section 3.1). The results also showed that no keto group was present on the β-ring on the left side of the carotene backbone (Fig. 1), confirming the deletion of the crtO gene. Carotenoids with a linear ψ-end and a β-ring have the exact same mass and the deletion of the crtY gene could therefore not be confirmed by mass spectrometry alone. However, the change from a β-ring and ψ-end should increase the wavelength of the absorption spectrum, due to the lower contribution of the β-ring to the bathochromic shift (Krinsky et al., 2004). The absorption spectra of the carotenoids extracted from ISCaR-493 and TK-1 (Fig. 3) show a clear shift towards higher wavelengths for the TK-1 carotenoids, suggesting the presence of a ψ-end and therefore the deletion of the crtY gene.
The extract of R. marinus strain TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) showed a sole peak (tR 2.75 min) by DAD (λ450 nm). This peak had a mass (m/z 536.428 ± 0.010) corresponding to C40H56 carotenoids, such as α-carotene, β-carotene, γ-carotene and lycopene. By comparing the tR of the peak of TK-3 to these carotene standards, TK-3 had the same tR as lycopene (Figs. S12 and S13). In addition, MS/MS fragmentation spectra were analyzed. The carotenes γ-carotene and lycopene have acyclic ψ-ends, which can be determined by removal of an isoprene unit, resulting in a [M-69]+• fragment of m/z 467.368 (van Breemen et al., 2012). This fragment could be detected in the standards γ-carotene and lycopene but also in the TK-3 MS/MS spectrum at tR 2.75 min. Moreover, absorption spectra of the TK-3 extract (Fig. 3) shows an identical wavelength profile as established spectra for lycopene (Britton et al., 2004). From these results it can be concluded that lycopene is produced by TK-3 as the sole product from the carotenoid biosynthetic pathway. Lycopene was quantified spectrophotometrically using Beer’s law in TK-3 extracts in a separate shake flask experiment. The result was a yield of 0.49 ± 0.01 g/kg cell dry weight (CDW), which corresponded to 0.14 ± 0.004 mg/L cultivation volume. In conclusion, the results suggest that the cruF-crtB gene was successfully deleted from the genome of R. marinus and that the heterologous crtB gene from T. thermophilus encodes an active enzyme in strain TK-3. Additionally, and somewhat surprisingly, the only active carotenoid enzymes in TK-3 seem to be CrtB and CrtI, leaving the other enzymes, such as CrtY, non-functional.
4. Discussion
Three successful modifications were performed in the carotenoid gene cluster of R. marinus, resulting in two carotenoid producing mutant strains. Based on the results obtained here, we propose the carotenoid biosynthesis pathway in R. marinus, which enzymes are involved and in what order they act. The mutant TK-1 (ΔtrpBΔpurAΔcrtYO::trpB) produces a lycopene backbone with all native modifications (displayed on the right side in Fig. 1) still present, but without the 4-keto β-ionone ring. The carotenoid genes crtY and crtO, which encode the enzymes that catalyze the keto ionone ring formation, were a part of the 5890 bp region from the larger operon that was deleted in TK-1. The remaining genes of the region apparently do not play a role in the biosynthetic pathway. The enzymes responsible for the modifications displayed on the right side of the backbone are mostly encoded by genes located in the smaller operon of the gene cluster (Fig. 2). They are active without the keto ionone ring according to the structure analysis of the carotenoids obtained from TK-1. This means they are not dependent on the activity of CrtY and CrtO.
The results indicate that the mutant strain TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus) produces lycopene as the sole carotenoid. This mutant has the cruF-crtB gene (fused cruF and crtB genes) deleted from its genome and the crtB gene from T. thermophilus inserted. Deleting only the cruF part of the gene did not result in a carotenoid producing strain (TK-2). Apparently, the crtB part by itself does not result in the corresponding enzyme activity that is high enough to produce carotenoids at detectable levels. Fused carotenoid genes have been observed in other species, such as the crtYB gene in Xanthophyllomyces dendrorhous. Deletions in the lycopene cyclase domain of this gene did not result in an active phytoene synthase either (Xie et al., 2015). TK-3, however, produces lycopene, implying that the crtB gene from T. thermophilus encodes an active enzyme in R. marinusand also that without CruF activity, most of the remaining enzymes downstream in the pathway cannot act on the carotene backbone. The CrtI enzyme is clearly still active, as it is essential for producing lycopene. This means that CrtI is not dependent on the modification done by CruF and that the two enzymes might act simultaneously in the native pathway. Since cruF and crtB are fused in one gene, it can be argued that their corresponding enzymes likely act together or successively. Whether the enzymes are fused into one polypeptide chain or not cannot be concluded from the data presented here. However, our results do show that CrtB is inactive without CruF, which suggests that the two enzymes might be acting as one entity. Also, coupled CruF and CrtB activities could explain how the organism is able to produce asymmetric carotenoids. While we can only speculate on the order of which these enzymes act, further studies on substrate specificity of the enzymes could elucidate their respective functions. The CruF enzyme is likely a 1′,2′-hydratase. Without its activity, it was to be expected that the CruC and CruD enzymes, which add a glycosyl and an acyl group to the C-1′ hydroxyl group, would not modify the backbone. Our results confirmed this. Two additional native modifications on the right side of the backbone were absent in the carotenoid from TK-3, which are catalyzed by unknown enzymes in R. marinus. They add the C-2′ hydroxyl group and the C-3′,4′ double bond. CrtI is a desaturase and has previously been reported to produce 3,4-didehydrolycopene in Neurospora crassa (Hausmann and Sandmann, 2000). It is therefore possible that the CrtI in R. marinus is responsible for the 3′,4′-desaturation. However, since the C-3′,4′ double bond is not present in TK-3, this putative activity of CrtI must be dependent on the hydration of the C-1′,2′ double bond. While we do not have stronger evidence of CrtI catalyzing the 3′,4′-desaturation in R. marinus, this step in the pathway remains unknown (Fig. 1). On the left side of the native carotene backbone is a 4-keto β-ionone ring, which is not present in the carotenoid produced in TK-3. The crtY gene, which encodes a lycopene cyclase, was not disrupted during the genetic modifications, which means that the corresponding enzyme simply cannot act on the backbone without the modifications on the right side of it. Based on this, the enzymes responsible for the modifications on the right side are proposed to modify the backbone before CrtY and CrtO (Fig. 1). To confirm this hypothesis, substrate specificity of the enzymes would have to be tested.
The objective of this work was to engineer the carotenoid biosynthetic pathway in R. marinus to produce lycopene instead of its native carotenoids. We successfully obtained the R. marinus mutant strain TK-3 (ΔtrpBΔpurAΔcruFcrtB::trpBcrtBT.thermophilus), that produced 0.49 g/kg CDW of lycopene. This can be compared to optimized commercial lycopene producing microorganisms, such as Escherichia coli, Saccharomyces cerevisiae and Blakeslea trispora, which have been reported to produce 43.7 (Zhu et al., 2015), 55.6 (Chen et al., 2016) and 15 (Hu et al., 2013; Tereshina et al., 2002) g/kg CDW of lycopene, respectively. Currently, R. marinus produces 1-2 orders of magnitude less lycopene than these strains. However, there is considerable room for improvement. Further optimization of the carotenoid production through genetic engineering and culture conditions is likely to improve yields significantly. For instance, in previous work on cultivation of R. marinus, a 28-fold increase of 450 nm absorption was observed in native carotenoid extracts from sequential batch cultivation with cell recycling, as compared to that of shake flask cultivations (Ron et al., 2019). Taking the higher cell densities into consideration, carotenoid absorption per cell density was still 11-fold higher than shake flask cultivations.
R. marinus is a robust versatile organism and in many aspects a preadapted production organism for utilization of recalcitrant polysaccharides in 2nd and 3rd generation biomass, an important task of biorefinery development. In the present study we have demonstrated that R. marinus is amenable to metabolic engineering, comprising both gene deletions and insertions, leading to efficient production of a metabolite of industrial interest. The work also revealed the potential of the methodology to help unravel complex pathways.
Declaration of competing interest
None.
Funding
This work was supported by the Marine Biotechnology ERA-NET, ThermoFactories, project grant number 5178–00003B; the Technology Development fund in Iceland, grant number 159004-0612; the Icelandic Research fund, ThermoExplore, project grant number 207088-051 and the Novo Nordisk Foundation (NNF18OC0034792). ENK, CT, EYCR and DM-D would like to acknowledge the Swedish Research Council Formas (2018-01863).