1. Introduction
Fluorescence in situ hybridization is a technique that is used for the spatial detection and quantification of nucleic acids in their cellular environment. It has emerged as a powerful cytogenetic method for the analysis of cells and tissues on the transcriptome and genome level with more than 6 publications in the literature per day since the late 1990s (Fig. 1a). In diagnostics, FISH is considered the gold standard cytogenetic method for the detection of diseased or malignant cells containing chromosomal rearrangements [1] or gene aberrations [2,3]. From its development in the 1960s onwards, FISH largely benefitted from improvements of probe-labeling techniques and specific probe design strategies increasing its sensitivity. Its broad application in research and diagnostics shows in the steep rise in the number of publications reporting FISH beginning in the early 1990s (Fig. 1a). At the same time, microfluidic techniques were first mentioned in the literature. The better understanding of fluid mechanics at the micrometer length scale has led to the convergence of microfluidics and biochemical assays, enabling their miniaturization. The first reports combining FISH and microfluidic approaches appeared in parallel with a large increase in the number of microfluidic applications from 2003 onwards. In recent years, the combination of microfluidic techniques and FISH addresses limitations in probe consumption and hybridization times, making the experimental procedure more sustainable and adaptable to high-throughput developments.
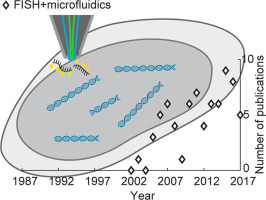
 Fig. 1. In situ hybridization – frequency of usage and implementation. (a) Number of publications containing keywords for FISH and microfluidics over the past 30 years. The number of publications per year is displayed based on searching the PubMed database for the sum of the keywords “fluorescence in situ hybridization” and “fluorescence in situ hybridisation” (black open squares), the keyword “microfluidic*” (black open circles), and the sum of the keywords “fluorescence in situ hybridization” AND “microfluidic*” and “fluorescence in situ hybridisation” AND “microfluidic*” (orange open diamonds). (b) In situhybridization for visualization of chromosomal targets. Labeled DNA probes (yellow) enter into permeabilized cells and nuclei and hybridize to their complementary target DNA sequences. In regions where labeled probes have hybridized to the target DNA, an ISH signal can be detected.
Fig. 1. In situ hybridization – frequency of usage and implementation. (a) Number of publications containing keywords for FISH and microfluidics over the past 30 years. The number of publications per year is displayed based on searching the PubMed database for the sum of the keywords “fluorescence in situ hybridization” and “fluorescence in situ hybridisation” (black open squares), the keyword “microfluidic*” (black open circles), and the sum of the keywords “fluorescence in situ hybridization” AND “microfluidic*” and “fluorescence in situ hybridisation” AND “microfluidic*” (orange open diamonds). (b) In situhybridization for visualization of chromosomal targets. Labeled DNA probes (yellow) enter into permeabilized cells and nuclei and hybridize to their complementary target DNA sequences. In regions where labeled probes have hybridized to the target DNA, an ISH signal can be detected.While the number of publications making use of FISH is slowly reaching a steady state, the number of microfluidic developments and their application to FISH is rising to this date. We speculate this is due to the rapid developments and advancements in the microfluidics field, in particular related to biomedical applications. We expect that microfluidics can largely influence the experimental design of FISH implementations and address some of its limitations in the coming years. In this perspective article, we revise some historical aspects in the development of FISH and highlight selected implementations of microfluidics-based FISH.
1.1. In situ hybridization
In situ hybridization (ISH) is a cytogenetic technique allowing high-resolution detection, quantification, and localization of nucleic acid targets inside cells or tissues. The method is based on the hybridization of sequence-specific complementary probes (typically DNA sequences) to their target inside the cell. Fig. 1b shows schematically how a fluorescently-labeled nucleic acid probe enters the cell nucleus where it hybridizes to its complementary target in the DNA. Probes can be targeted at DNA (metaphase and interphase chromosomes) as well as RNA, making the study of genomic sequences and transcriptomic expression profiles of individual cells possible. The reaction can be visualized directly using radioactively or fluorescently labeled probes or indirectly viahistochemical chromogens, antigen binding, or biotin-streptavidin interactions.
Binding of complementary probes to the target of interest thus enables its detection and visualization. This visualization can be leveraged for medical applications such as prenatal diagnostics or oncology. Using probes targeted to different chromosomes or chromosomal regions (chromosome painting), chromosomal aberrations such as differences in enumeration, translocations, insertions, or deletions can be analyzed [4,5]. Some examples of diseases diagnosed using ISH include cystic fibrosis, Duchenne's muscular dystrophy, Prader-Willi syndrome, Angelman syndrome, Lejeune's syndrome, trisomies 13, 16, 18, 21, monosomy X, Turner syndrome, and different types of leukemia. Furthermore, gene-specific targets can be used to detect gene amplifications often found in tumor cells. An example is the detection of human epidermal growth factor receptor 2 (HER2) gene copy number changes as prognostic and predictive biomarkers in breast [6] and ovarian cancer [7,8]. ISH additionally plays a role in the species identification of (pathogenic) microbes [9].
Therefore, ISH is a common tool in cell biological research as well as in diagnostic applications, as it provides valuable quantitative information about the nucleic acid distribution inside individual cells. A large advantage of the technique is its ability to provide spatial information of cellular content, thus providing the possibility of investigating spatial heterogeneities.
1.2. A historical view on in situ hybridization
ISH was developed and matured much later than immunofluorescence-based protein detection methods. While immunofluorescence was first used by Coons et al. [10] in 1941 and started to be applied regularly in the 1950s, in situhybridization was introduced ~30 years later (Fig. 2). Melting and re-hybridization of DNA and complementary RNA-DNA binding were first described in the 1960s. In 1969, Pardue and Gall then tagged rRNA probes with H3 to autoradiographically visualize rRNA-encoding genes in cytological preparations of Xenopus laevis oocytes [11]. In parallel, John et al. [12] and Buongiorno-Nardelli and Amaldi [13] independently developed in situhybridization techniques for the detection of rDNA in Xenopus and paraffin-embedded tissues. A few years later, Gall et al. visualized satellite elements in heterochromatic regions of mouse chromosomes [14,15] demonstrating the use of ISH in mammalian cells. Limitations of these early ISH experiments were the low sensitivity and the limited availability of sequence-specific probes. Although radioisotope labeling is still considered the most sensitive method of detection, it requires long exposure times (up to weeks for the detection of H3), the spatial resolution is low (in the range of Mega bases), and the background levels are high [11]. These disadvantages led to the development of non-isotopic labeling approaches for ISH probes.
 Fig. 2. Timeline of fluorescence in situ hybridization developments. The earliest record of in situ hybridization is found by Gall and Pardue in 1969 [11]. First fluorescent versions of the technique (FISH) appeared in the 1970s, followed by direct probe labeling twenty years later. ‘Modern’ FISH further includes developments in the probe design and production as well as background reduction. The combination of microfluidics and FISH first appeared in the early 21st century. Events directly related to the development of FISH are shown in boxes, while other findings influencing FISH are displayed as text only [118], [119], [120].
Fig. 2. Timeline of fluorescence in situ hybridization developments. The earliest record of in situ hybridization is found by Gall and Pardue in 1969 [11]. First fluorescent versions of the technique (FISH) appeared in the 1970s, followed by direct probe labeling twenty years later. ‘Modern’ FISH further includes developments in the probe design and production as well as background reduction. The combination of microfluidics and FISH first appeared in the early 21st century. Events directly related to the development of FISH are shown in boxes, while other findings influencing FISH are displayed as text only [118], [119], [120].1.2.1. From radioactive to fluorescent probe labels
In the mid-1970s, a range of non-radioisotopic probes were developed, allowing for new detection options of the probe-target hybrids. In one approach, a poly(rA)-poly(dT) antibody was used to detect RNA-DNA hybrids on Drosophilachromosomes [16]. The signal was created using a rhodamine-conjugated secondary antibody. Others used rRNA-biotin, which was then indirectly detected via an avidin-based detection system, and visualized using a scanning electron microscope [17]. Langer-Safer and Ward (1982) [18] on the other hand used nick-translation to incorporate biotin-labeled bases into DNA, which were then detected by primary and fluorescently- or enzymatically-conjugated secondary antibodies.
Direct fluorescent detection of chromosomal targets was reported by Bauman et al. in the 1980s, who labeled the 3′ ends of the probes with a fluorochrome [19]. The combination of direct fluorophore-labeling of nucleic acids and enzymatic incorporation of fluorophore-modified bases from these publications [18,19] lay the ground for the facilitated preparation of fluorescent probes. Probes with fluorescein-labeled bases incorporated using nick-translation showed detection sensitivities of 50–100 kb [20] and were used for first multicolor FISH experiments. The development of amino-allyl modified bases [21] for the conjugation with haptens or fluorophores further facilitated the production of a large variety of FISH probes. Owing to sparse labeling of long probe sequences for specific hybridization, FISH was still limited by the detection sensitivity. Indirect detection using secondary reporter systems based on biotin-avidin interactions [22] or antibody binding [18] could amplify the generated signal.
The chemical synthesis of DNA probes finally allowed the incorporation of enough fluorophores for direct detection [23]. A range of different probe labeling techniques followed for generating densely labeled FISH probes, such as digoxigenin labeling of the probe [23], cis‑platinum complex-mediated labeling [24] or polymerase chain reaction (PCR) [25] and degenerate oligonucleotide primed PCR [26], among many others [27,28]. This enabled efficient generation of probes carrying various labels for multiplex FISH [[29], [30], [31], [32]], which led to new methods such as comparative genomic hybridization (CGH) [33] for cytogenetic analysis of tumors, or spectral karyotyping by chromosome painting [34,35]. Recent developments include the use of quantum dots [36,37] and click chemistry [38].
The commercialization of fluorescence microscopes beginning in the 1970s and the developments in the fields of confocal or multiphoton microscopy in parallel to the design of new labeling schemes paved the way for highly sensitive detection of FISH signals.
1.2.2. FISH probe design
From the early developments of ISH to the late 1970s, polytene chromosomes or satellite sequences were the ISH targets of choice, which, owing to their repetitive sequences, enabled ISH signal detection in the absence of sensitive probes or detectors. With the development of gene cloning and new probe labeling strategies, the specific design of FISH probes became possible improving their specificity significantly. While first probes were generated by isolation of crude RNA to target large fragments of chromosomes in the late 1980s, gene-specific FISH probes could be generated from gene libraries from 1986 onwards [39]. However, libraries generated from microdissected chromosomes contain repetitive elements, which are spread across the genome. Thus, FISH probes had the tendency to not only hybridize to the specific target but also to off-target sequences in the cell. Methods for masking such repetitive elements with salmon sperm or Cot-1 deoxyribonucleic acids (DNA) were developed [40], and recently, Swennehuis et al. reported a method for enzymatic removal of repetitive elements to create unique, repeat-free FISH probes from artificial chromosomes [41].
Thanks to advances in the Human Genome Project [42,43], bioinformatics tools [44] and oligonucleotide synthesis [45], synthetic FISH probes are now being increasingly used as they can be easily designed and synthesized. The use of oligonucleotide libraries as a source for probes offers the possibility of detecting chromosomal regions in the range of tens of kilobases to megabases [46]. Bioinformatic pipelines [[46], [47], [48]] for the selective design and de novosynthesis of oligonucleotides facilitate the development of chromosome-specific painting probes. Synthetic oligonucleotide probes are particularly powerful, as they can be specifically designed to be sensitive enough to detect splicing variants [49,50] and even single nucleotide polymorphisms [51,52]. Lately, in situ hybridization-based single-cell RNA sequencing approaches were developed using oligonucleotide probes flanked with primers for rolling circle amplification [53,54]. In addition to synthetic oligonucleotide-based FISH probes, also DNA analogous probes such as peptide nucleic acid and locked nucleic acid probes are regularly used in FISH experiments [55,56]. As, in contrast to DNA, they do not contain a charged backbone, these nucleic acid analogues hybridize more efficiently and specifically than DNA owing to minimal electrostatic interactions with the sample.
The advances in the field of probe design and labeling strategies allowed FISH to become a highly versatile technique addressing a broad range of cellular targets with high specificity and sensitivity. FISH could thus be used to enumerate chromosomes, detect chromosomal aberrations, investigate specific chromosomal regions or transcriptional levels. These aspects are not only interesting for cytogenetic research applications, but additionally provide the basis for molecular diagnostics in a range of medical fields.
2. FISH hybridization kinetics
A key aspect of the FISH assay is the hybridization of the probe to its target. The hybridization reaction of the free probes to chromosomal targets in the nucleus can be described using second-order reaction kinetics [57],(1)where cP is the concentration of the FISH probe, cT is the concentration of target in the nucleus, cPT is the concentration of bound probes, and kon and koff are the on- and the off-rates of the hybridization reaction, respectively.
With the assumption that cP > > cT, the characteristic hybridization time of a FISH reaction can be defined as(2)
Thus, a high concentration of FISH probes in the nucleus enhances the hybridization rate. Standard FISH reactions are typically performed using a bench-top assay by pipetting the FISH hybridization mix onto the cytological sample. The diffusion-based transport of FISH probes in solution and in the cell is slow, and the diffusion coefficient decreases with increasing probe length [58,59]. This leads to typical incubation times of up to 16 h for low copy number targets [60,61] and 48 to 94 h for entire genome hybridization [62].
This rate-limiting step can be overcome by performing a flow-based FISH probe incubation step, resulting in a convection-enhanced transport of FISH probes into the cells and thus an increased FISH probe concentration cP in the cell. Thus, the hybridization time and with this the assay time can be reduced using microfluidic devices employing an active delivery of FISH probes to the cells, as demonstrated by the microfluidic community [[63], [64], [65]]. The microfluidic delivery ensures a constant replenishment of probes on the surface of the permeabilized cells and tissues. The reaction is therefore limited by the diffusion inside the tissue and the hybridization reaction.
3. Conventional implementations of FISH
A conventional FISH reaction is typically performed as a bench-top assay by pipetting the FISH hybridization mix, containing the probes, onto the cytological sample and incubating it (Fig. 3). FISH can be performed on suspended cells followed by cell sorting procedures for separating the fluorescent signal as well as on cultured cells and frozen or formalin-fixed paraffin-embedded tissue sections. During fixation of the sample, it is important to preserve nucleic acid integrity and cell morphology. The actual experimental FISH procedure includes several preparatory steps, the hybridization reaction itself, and the removal of unbound probes. A short outline of the procedure is illustrated below.
 Fig. 3. Schematic representation of the experimental FISH assay procedure. The sample of interest is fixed and pretreated to facilitate the penetration of FISH probes. A potential denaturation step is followed by hybridization with the FISH probes under stringent buffer and temperature conditions. Unbound probes are removed by washing to ensure imaging of specifically stained regions only. Counterstaining of specific cellular structures aids in cell selection. Signal detection is performed on a fluorescence microscope followed by a qualitative or quantitative analysis.
Fig. 3. Schematic representation of the experimental FISH assay procedure. The sample of interest is fixed and pretreated to facilitate the penetration of FISH probes. A potential denaturation step is followed by hybridization with the FISH probes under stringent buffer and temperature conditions. Unbound probes are removed by washing to ensure imaging of specifically stained regions only. Counterstaining of specific cellular structures aids in cell selection. Signal detection is performed on a fluorescence microscope followed by a qualitative or quantitative analysis.3.1. Experimental procedure
Depending on the sample of interest, pretreatment of the sample can be performed to facilitate the entry of FISH probes and thus increase their hybridization efficiency. In the case of cultured cells, these typically include fixation and cell permeabilization using organic solvents or formaldehyde. For tissue sections, formalin cross-linking might have to be removed before subsequent protease treatment revealing the target sequences and improving FISH probe penetration into the tissue.
The actual hybridization reaction then takes place by incubating the FISH probes with the sample of interest at a temperature providing specificity for the hybridization reaction for several hours. The concentration of formamide in the hybridization buffer changes the stringency of the hybridization reaction and needs to be chosen probe-specifically. The buffer composition as well as the hybridization temperature determine the sensitivity and specificity of the reaction and are typically defined empirically based on the length of the probes and the sample of interest. Unspecific charge-charge interactions of the probe with the tissue can be reduced further using a mixture of acetic anhydride and triethanolamine.
Several stringent washing steps after the hybridization ensure removal of unbound FISH probes and specificity of the detected signal. Additionally, nuclear staining is often performed before FISH signal detection on an epifluorescent or confocal microscope or even using super-resolution microscopy. For high-quality images, a large magnification is used, which results in the collection of a large number of images with small fields of view.
3.2. Analysis
Following the detection and imaging of the fluorescent signal obtained from the hybridization reaction, a quantitative analysis of the results is often desired. To this day, the manual counting of FISH signals per cell or nucleus is still the standard approach, making the data analysis the post-experimental rate-limiting step. The analysis is based on counting individual sequences of interest, which are visualized by fluorophores attached to the hybridization probe. Diagnostic decisions are often based on determining the ratio of the number of signals from two distinct chromosomal regions to determine chromosomal or gene amplifications; an example is testing of the ratio HER2/CEP17 for the molecular characterization of breast cancer tissue [66]. A sufficient intensity for detection is achieved by combining several fluorophores on a single probe and by using sensitive cameras. A threshold is then applied to distinguish the FRETsignal from background autofluorescence.
Owing to the large number of high-resolution images obtained, several algorithms for the automatic detection and counting have been developed. These algorithms are based on the detection of the cells or stained nuclei in the interphase or metaphase configuration [67]. Masking of the detected geometries is then used to determine the regions of interest for dot-counting [[67], [68], [69]]. Table 1 presents a non-exhaustive overview of software and algorithms used for semi-automated and automated FISH image analysis.
Table 1. Software and algorithms for (semi-) automated FISH image analysis.
| Type of analysis | Software/algorithm |
|---|---|
| Automated cell-by-cell quantitative expression profile analysis |
|
| Semi-automated smFISH data analysis |
|
| Automated image processing (DNA FISH) |
|
| Semi-automated scoring (DNA FISH) |
|
4. Challenges of current FISH assays
Fluorescence in situ hybridization provides genomic and transcriptomic information in the spatial cellular context. Thanks to its unique advantages, it has found applications in cell biological and genomic research as well as clinical diagnostics in preventive and reproductive medicine and oncology. Especially in the context of clinical applications, however, a few key limitations of the conventional FISH procedure become apparent.
Tremendous developments have been achieved for multicolor FISH over the past years. Two types of combinatorial labeling procedures, spectral karyotyping [34] and M-FISH [71], are used to detect the FISH signal either based on the spectral properties or on the absence/presence of specific fluorophores. Both methods can be applied for the specific labeling of the complete set of 24 human chromosomes, detecting translocations, insertions, or deletions and other aberrations. Theoretically, using seven fluorophores, combinatorial labeling produces 127 different signatures [72]. Since these early applications, a range of multicolor FISH procedures have been developed [72,73]. Recently, MER-FISH has increased the number of differentially labeled targets to 1001 [74]. However, the number of detected targets is still far below the expected value of different transcripts (approx. 12,000 different RNAs) in a cell. By combining spectral with other methods of multiplexing, the number of detected targets might be increased even further in the future. Furthermore, other approaches to multiplexed labeling might enable the use of simple equipment rather than epifluorescent or confocal microscopes with eight or more sets of filters.
The experimental implementation of FISH is time-consuming and requires experienced personnel, therefore the development of validated protocols is expensive. Furthermore, the different steps in the experimental procedure are probe- and sample-specific [65] and have to be optimized for each set of conditions empirically. This already requires the use of a large number of probes and precious clinical samples. As many probes are not yet commercially available, a large number of research laboratories still produce their own FISH probes. The design, preparation, and labeling of these probes is still very labor-intensive. Even if probes can be chemically synthesized, depending on the nucleic acid length and fluorescent labels, they can be expensive, especially when a large number of probes is required. Reducing the probe volume and size of the tissue areas used for improving the experimental workflow or providing insight into the effects of different parameters on the hybridization reaction could therefore strongly influence the importance of FISH in research and diagnostics in the coming years.
As FISH is performed as an end-point analysis method, little is known about the hybridization kinetics of the probes inside cytological samples. For a high level of comparability between samples, FISH protocols thus have to be chosen such that saturation of the signal is reached. To ensure saturation, long hybridization times of the probes are required, making the procedure time-consuming. Developing novel FISH protocols based on probe-specific information about hybridization kinetics can reduce the hands-on time of experienced personnel. Furthermore, reducing hybridization times and increasing automation of the assay and thus minimizing labor time and improving comparability between samples and laboratories can significantly improve FISH pervasiveness in clinical diagnostics.
5. Microfluidic FISH implementations
In recent years, microfluidic technologies have emerged as powerful tools for studying cells [75]. The microfluidic community has developed microFISH implementations (μFISH) for studying cytological samples. The unique physical properties of laminar flow and the constant replenishment of reagents can be leveraged to solve some bottlenecks of FISH by reducing the cost per test, automating the assay [76,77], or simplifying the implementation [78] (Fig. 4, Fig. 5, Fig. 6). At the same time, implementing FISH assays using microfluidic devices is challenging, especially as elevated temperatures are often favorable for hybridization. Some of the developments therefore focus on individual steps of the assay, while others propose to perform the whole procedure on-chip.
 Fig. 4. Selected examples of μFISH devices used for analysis of suspended cells and microbes. (a) In a chemistrode [85], mobile plugs of droplets are created. These can be used for the separation and cultivation of bacteria or selective chemical reactions. (b) The μflowFISH [83] chip combines an on-chip FISH assay with flow cytometric separation and detection for the identification of bacterial species. (c) Device for the miniaturization of the FISH assay using capillary forces [97]. The analysis of hematopoietic stem cells in this device resulted in a 10-fold reduction of probe consumption. (d) Microfluidic chip made from cyclic olefin copolymer designed for increasing cell immobilization in the chamber for subsequent FISH analysis [94]. Using the chip, FISH can be performed in a fully automated fashion with a 10-fold reduction in probe consumption. (e) Chip and mask layout of the first fully automated microfluidic implementation of FISH (including reagent multiplexing valves, a reaction chamber, and peristaltic pumps) for the detection of X and Y chromosomes [76].
Fig. 4. Selected examples of μFISH devices used for analysis of suspended cells and microbes. (a) In a chemistrode [85], mobile plugs of droplets are created. These can be used for the separation and cultivation of bacteria or selective chemical reactions. (b) The μflowFISH [83] chip combines an on-chip FISH assay with flow cytometric separation and detection for the identification of bacterial species. (c) Device for the miniaturization of the FISH assay using capillary forces [97]. The analysis of hematopoietic stem cells in this device resulted in a 10-fold reduction of probe consumption. (d) Microfluidic chip made from cyclic olefin copolymer designed for increasing cell immobilization in the chamber for subsequent FISH analysis [94]. Using the chip, FISH can be performed in a fully automated fashion with a 10-fold reduction in probe consumption. (e) Chip and mask layout of the first fully automated microfluidic implementation of FISH (including reagent multiplexing valves, a reaction chamber, and peristaltic pumps) for the detection of X and Y chromosomes [76]. Fig. 5. Selected examples of μFISH devices used for the molecular analysis of cell monolayers and tissue sections. (a) First μFISH device for analysis of tissue section, which focused on automation of the assay [98]. The device is based on a microfluidic control module for fluid transport combined with an air chamber to introduce pressure differences. (b) Microfluidic processor chip made of Pyrex-Si with flow-based hybridization capacities used for the assessment of HER2 status in tissue sections [63,64]. Copyright obtained.
Fig. 5. Selected examples of μFISH devices used for the molecular analysis of cell monolayers and tissue sections. (a) First μFISH device for analysis of tissue section, which focused on automation of the assay [98]. The device is based on a microfluidic control module for fluid transport combined with an air chamber to introduce pressure differences. (b) Microfluidic processor chip made of Pyrex-Si with flow-based hybridization capacities used for the assessment of HER2 status in tissue sections [63,64]. Copyright obtained. Fig. 6. Micro-scale FISH using an MFP in the open-space [106]. (a) Schematic of the microfluidic probe technology. A microfluidic scanning probe made of Si-glass exposes selected cells to FISH probes for spatially localized hybridization. (b) μFISH of CEP7 and CEP17 on cultured MCF7 cells. (c) Micrographs of FFPEcell blocks (top) and FFPE breast tumor sections (bottom) after μFISH. FISH signals represent the HER2 gene (red) and centromere 17 (green) copy number in the cell nuclei stained with DAPI (blue).
Fig. 6. Micro-scale FISH using an MFP in the open-space [106]. (a) Schematic of the microfluidic probe technology. A microfluidic scanning probe made of Si-glass exposes selected cells to FISH probes for spatially localized hybridization. (b) μFISH of CEP7 and CEP17 on cultured MCF7 cells. (c) Micrographs of FFPEcell blocks (top) and FFPE breast tumor sections (bottom) after μFISH. FISH signals represent the HER2 gene (red) and centromere 17 (green) copy number in the cell nuclei stained with DAPI (blue).5.1. A short introduction to microfluidics
New developments in the microfabrication of devices, namely, photolithography, paved the way for the application of fabricated chips in the field of fluidics. A first construction of a device for microfluidic analyses is often ascribed to Terry et al. (1979) [79], which was followed by a range of so-called lab-on-a-chip devices triggered by the work by Manz and Harrison [80]. In general, microfluidics deals with the handling, control, and manipulation of small volumes of liquids or gases, which can be achieved in devices with geometrical constraints limiting the flow channel dimensions to sub-millimeter sizes. The small size in combination with unique physical phenomena at the micrometer scale, such as laminar flow [81], capillary forces [82], and high surface-to-volume ratio affecting heat dissemination and diffusion properties, provide several advantages for the application of microfluidic approaches to biochemical assays. These include low sample and reagent consumptions and potentially increased reaction speeds. Defined reaction areas further enable parallelization, multiplexing, automation, and the extension to high-throughput experimentation.
5.2. Micro-scale FISH of suspended cells and adherent cytological substrates
A range of μFISH-based implementations for the analysis of bacterial and mammalian cells in suspension were developed, and separation was achieved by flow cytometry [83], capillary electrophoresis [84], or the chemistrode [85]. Fig. 4a shows a schematic of the chemistrode, fabricated by soft lithography in PDMS with surface silanization and attached to a syringe system, in which mobile droplets are created for the isolation of individual bacteria. By combining two streams of liquids, the bacteria were fixed inside the droplets and then positioned separately on a glass slide prior to 16S rRNA probe hybridization to identify individual P. curdlanolyticus cells. In this approach, the microfluidic device was thus used only prior to the actual FISH procedure. Packard et al. (2012) [86], in contrast, introduced a microfluidic-based Förster resonance energy transfer (FRET) assisted FISH method on chip for the rapid identification of bacteria. While typically the identification of bacterial speciesis achieved using real-time PCR, whose total experimental procedure requires hours, this microfluidic FRET-ISH protocol can reduce the time until identification to less than 30 min [86]. The combination of a microfluidic chamber for FISH with channels for cytometric detection of individual cells [83] provided an on-chip implementation of the whole assay procedure, termed μflowFISH, for species identification. Fig. 4b shows the design of the microfluidic chip used for μflowFISH. The glass chip was treated by photolithography and chemical etching to form a FISH chamber separated by photopolymerized polyacrylamide membranes and channel cross structures for electrokinetic focusing. The chip was used for concentrating and mixing the microbial cells and the FISH probes in a cyclic fashion with six incubation cycles of five minutes each and washing of the sample post hybridization. Similarly, combining capillary electrophoresis with FISH resulted in the identification of Salmonella typhimurium [84]. Combining on-chip FISH with immunofluorescence detection further increased the sensitivity of bacterial identification, while still providing short assay times of around 30 min [87]. Microscale FISH approaches provide several advantages over standard methods for bacterial identification, mainly by decreasing assay times and by using separation techniques for improving the single cell sensitivity and enabling the analysis of low numbers of cells.
μFISH-based approaches were also used for the detection of chromosomal translocations on metaphase spreads of mammalian cells in suspension [88,89]. By performing microfluidic-based chromosomal analysis, the throughput could be increased significantly while the reagent consumption was reduced [78]. These chromosomal analyses and cytogenetic tests play an important role in diagnosis. The miniaturization of FISH assays for diagnostics is based primarily on closed microfluidic channels and reservoirs to enable automation and decrease interlaboratory variation. Examples of these microfluidic chips are displayed in Fig. 4c-e. The devices were applied for prenatal diagnostics [90], the analysis of circulating tumor cells [[91], [92], [93], [94]], and cancer malignancies [[95], [96], [97]] using micro-scale FISH. A simple miniaturization of the FISH assay to a channel format in a chip fabricated from ns-TiO2functionalized glass reversibly adherent to PDMS-containing microchannels(Fig. 4c) already reduced the probe consumption 10-fold [97] by limiting the assay to the size of the channel. Cells in PBS were incubated in the flow chamber for adhesion and then fixed inside the chamber. Washing, protease digestion, denaturation, probe hybridization, and final washing were performed inside the chip, which was placed on a hot plate for temperature control. An automated microfluidic procedure with a similar reduction in probe consumption was applied to the detection of HER2 status of cells cultured on the chip surface [94]. To enhance the use of miniaturized FISH assays in the clinics, the chip was designed in a monolithic, disposable form (Fig. 4d). To this end, the structure containing wide and narrow microfluidic channels was introduced into cyclic olefin copolymer by hot embossing, and bonding was achieved by solvent-assisted plasticizing. Another fully automated microfluidic implementation of FISH was described by Sieben et al. (2008) [76], who used a microchip design made of three layers, glass/PDMS/glass, which includes bus valves to combine and mix reagents, a chamber for FISH hybridization heated by a thin-film heater, and a peristaltic pump system (Fig. 4e). The complete FISH protocol, including delivery and immobilization of cells, protein digestion, fixation, denaturation, probe delivery and hybridization, washing, and counterstaining, was automated on-chip requiring an assay time of 93 min and a hands-on time of only 5 min. Using this device for the detection of X and Y chromosomes, the group was thus able to reduce the reagent consumption of the FISH assay by 20-fold and decrease the labor time significantly owing to automated hybridization.
The automation of FISH reduces labor time and variability in the experimental procedure, parameters which are highly important for clinical applications. Furthermore, they provide a more cost-effective alternative to conventional FISH assays. Owing to these advantages, sensitive micro-scale FISH procedures will find broad applications in the clinical setting, ranging from the detection and identification of prokaryotes to the analysis of cytogenetic information in circulating tumor cells and hematological malignancies.
While the handling of suspended cells in microfluidic devices has been successfully applied in the clinics, the implementation of microfluidics to on-slide cytological preparations or tissue sections poses the additional challenge of keeping the sample intact during the assay procedure. For μFISH-based analysis of cell monolayers and tissue sections, Kao et al. focused on automating the complete FISH assay using static hybridization [98]. They fabricated a chip consisting of three layers: an upper PDMS air chamber (1)with a membrane on top of a liquid PDMS chamber (2), produced by PMMA and PDMS molding, and a glass slide (3)(Fig. 5a). Fluids were transported inside the liquid chamber by pressure differences in the air chamber. The group applied their approach to the detection of HER2 amplification within timescales of 20 h and with a 70% reduction in reagent consumption. Nguyen et al. [63,64] introduced the flow-based incubation of FISH probes for the assessment of the HER2 status of tissue sections. Fig. 5b shows the chip used for these flow-based hybridizations, consisting of a Pyrex-Si stack with branched channels etched into the Si-layer to achieve homogeneous distribution of the reagents on the tissue. The microfluidic chamber was formed by mechanical clamping of the chip to the sample of interest. A copper holder placed on a hot plate was used for temperature control during the reaction. The denaturation and hybridization steps of the FISH protocol were performed within the chip. Flow-based incubation resulted in convective delivery of probes, reducing the hybridization time to approximately 35 min to 2 h, depending on the type of hybridization buffer used. For RNA-FISH in tissue sections, the Histo Flex [99] method, based on an elastomeric lid patterned with microfluidic channels, used flow-based hybridization to implement the localized and multiplexed detection of 18S rRNA and microRNAs.
While providing large improvements in terms of probe consumption and hybridization time compared to conventional FISH, these microfluidic devices come into direct contact with the sample. This physical contact may easily damage the fragile cytological sample. Furthermore, the geometry of the microchannels is fixed, whereas histological samples can be heterogeneous in terms of morphology.
5.3. Micro-scale FISH in the ‘open space’
There is an emerging trend in the microfluidics community towards the so-called ‘open-space’ microfluidics, enabling analysis on biological substrates without physical contact between the (cytological) sample and the microdevice. Open-space microfluidics include droplet microfluidics and microreactors that are open to the environment. Examples of open-space technologies are the chemistrode [100], multifunctional pipette [101], fluid force microscope [102], single-cell pipette [103], or the microfluidic probe (MFP) [104,105]. These technologies enable (bio)chemical analysis of ‘native’ substrate at the micrometer length-scale, without the need for its immobilization within the microfluidic channels. We focus here on the MFP and its variants. The MFP is a non-contact scanning probe technology and localizes reagents on selected areas of immersed substrates at the micrometer length-scale. It is thus a technology without constraints to the substrate, which could include Petri dishes, tissue sections, cell blocks, or any specimen immobilized on a surface. The microfluidic chip is the 'head' of the MFP containing a Si layer with microfabricated structures bonded to glass using photolithography and deep reactive ion etching. A hydrodynamic confinement of liquids on the sample surface is achieved by reagent flow between two apertures localized at the tip of the MFP, which is in proximity to the substrate such that the probe apex is positioned 10s of micrometer away from the immersed surface (Fig. 6a).
Using this technology, an MFP-based micro-scale FISH method was developed [65]. The procedure was based on conventional FISH implementations for its preparatory (fixation, permeabilization) and its visualization (washing, counterstaining) steps. Using this approach for FISH-based chromosomal enumeration in cell monolayers, the hybridization time was reduced to 10 min and the probe consumption to 2 μL [65]. The method allows spatially multiplexed FISH analysis to be performed on a single sample using probes targeting different chromosomal regions (Fig. 6b). Furthermore, the method is compatible with HER2 assessment of formalin-fixed paraffin-embedded (FFPE) breast tumor samples with 15 min incubation time and a probe consumption of 107 nL per test, which is a ~100-fold reduction compared to the standard protocol [106] (Fig. 6c). Besides its ability to address some key limitations of the FISH assay, the great advantage of using a non-contact microfluidic technology for FISH analysis is the absence of physical contact between the sample and the device, avoiding histological and morphological artifacts.
5.4. Micro-scale FISH as a tool for the rational design of FISH assays
In recent years, there is growing interest in a more precise evaluation of in situhybridization rates to achieve probe designs with improved sensitivity and specificity and shorten the typically long assay times [107]. A range of mathematical models was proposed to predict the hybridization kinetics of FISH reactions [[108], [109], [110], [111]]. However, despite their fundamental importance to FISH assay development, there are no techniques available, to this date, for the quantification of hybridization rates that can be readily applied to pairs of probes and targets. Thus the design procedures of FISH probes and assays remain largely empirical, and hybridization conditions for a given probe are estimated experimentally using an end-point analysis of the signal [107,108]. This remains a highly complex, labor-intensive and time-consuming task when multiple effects and conditions are to be tested.
MFP-based methods, however, allow FISH reactions to be monitored in real-time and subsequently enable measurements of the FISH hybridization kinetics [112]. Intracellular hybridization kinetics are measured in real-time by rapid switching between localized delivery of a fluorescent probe solution and a non-fluorescent imaging buffer. Fig. 7a shows the observed intensity profile of FISH signals developing over the time of hybridization. The extracted probe binding rates (Fig. 7b-c) can be used to determine optimal hybridization conditions. Such real-time monitoring of FISH assays allows effects of various buffer components, temperature, and the probes themselves on the hybridization rate to be investigated quantitatively and thus presents a valuable tool to develop models for the rational design of FISH assays.
 Fig. 7. Analysis of FISH probe hybridization during micro-scale FISH using an MFP [112]. Experimental results showing kinetic measurements of FISH for CEP17 probes under different hybridization conditions. (a) Normalized intensity during the hybridization reaction over time for different buffer compositions. (b) Corresponding FISH images after hybridization of CEP17 probes in buffers with ionic strengths ranging from 150 mM to 1 M. (c) Hybridization on-rate dependent on the ionic strength of the buffer.
Fig. 7. Analysis of FISH probe hybridization during micro-scale FISH using an MFP [112]. Experimental results showing kinetic measurements of FISH for CEP17 probes under different hybridization conditions. (a) Normalized intensity during the hybridization reaction over time for different buffer compositions. (b) Corresponding FISH images after hybridization of CEP17 probes in buffers with ionic strengths ranging from 150 mM to 1 M. (c) Hybridization on-rate dependent on the ionic strength of the buffer.6. Conclusions and outlook
Since its implementation in the 1960s, FISH has grown to be an important, powerful and sensitive technique for the detection of chromosomal abnormalities and changes in gene expression levels in microbes, cells and tissues for research and diagnostic applications. Although researchers have taken advantage of the possibilities FISH experiments offer, such as the high sensitivity in the spatial detection of specific genomic and transcriptomic sequences, a few challenges need to be overcome to exploit the full potential of FISH, especially for diagnostic applications.
Microfluidic implementations of FISH address some of the key limitations, such as probe consumption and hybridization time. We expect that by reducing the hybridization and thus the assay time and simultaneously lowering the economic footprints thanks to reduced probe cost, the frequency of FISH applications mainly in the diagnostic setting will increase significantly. Great advances in the field of micro-scale FISH were achieved using open-space microfluidics leveraging the advantages of microfluidics in a non-contact manner. While addressing the above-mentioned limitations of FISH, the cellular and histological morphology is unaffected. This facilitates the design of FISH probes and the development of automation procedures for micro-scale FISH implementations as avoiding mechanical contact between the device and the sample diminishes the risk of sample damage during the process, rendering high-throughput approaches possible.
Using micro-scale FISH, spatial multiplexing of different probes on a single tissue is possible. This offers the possibility of detecting multiple target sequences using a single dye and detection scheme, which makes FISH utilizable even in laboratories with less extended microscopic facilities. Furthermore, specific probes can be chosen for the hybridization in specific regions of a single heterogeneous sample, reducing the use of precious clinical sample. Besides reducing the FISH detection to a single colour, the combination of spatially and spectrally multiplexed FISH [113] can lead to the detection of even more specific sequences simultaneously. This might be of special interest on the transcriptomic level, where the current spectral combinations are not sufficient to detect all the different RNAs or splice variants in a single cell. The combination of MER-FISH approaches [74] with the spatial separation of sequence-specific detection using microfluidic devices could enable the investigation of large parts of the entire transcriptome with the advantage of additionally providing spatial information.
Furthermore, open-space microfluidic devices enable real-time monitoring of the hybridization kinetics of FISH probes [112]. Investigations of these kinetics will enable fine-tuning of the assay parameters for the hybridization reaction, such as buffer components, ionic strength, formamide concentration, and temperature. By scaling the assay to micrometer lengthscales, sequential testing of a multitude of parameters on a single cytological preparation with low probe consumption becomes possible. The optimization of experimental procedures will make the FISH assay more robust and user-independent. Apart from making FISH more stable, these new implementations can aid in characterizing probe specificity and sensitivity and in combining it with mathematical models. By continuous feedback from experimental results and analytical models, eventually an algorithm could be generated to design FISH probes with certain specificity, sensitivity, and hybridization conditions. The understanding of the fundamental biochemical reactions during FISH probe hybridization could therefore revolutionize the ways new FISH probes and experimental procedures are designed.
With the advent of whole genome and single-cell sequencing approaches, the use of FISH for genomic and transcriptomic analyses has certainly been affected. However, its unique ability of providing spatial information in combination with gene or transcript information plays an important role in its future use. Several in situ sequencing techniques have been described in the literature [53,74,114]. We believe that these might benefit from microfluidic applications, which could enable the local amplification or reverse transcription of specific sequences. A localized sequence-specific amplification of different targets might enable the detection of low copy number genes and transcripts which is a challenge to this day for FISH as well as for next generation sequencing applications. Using micro-scale approaches for the local amplification of targets by in situ labeling [115] or in situ transcription and fluorescence in situsequencing [116] would enable the detection of several genes or transcripts with low expression in different spatial locations on the same sample.
We can imagine that the new scientific findings on probe hybridization kinetics resulting from micro-scale FISH will not only have implications for the FISH assay itself, but they can be extended to a range of related techniques. For example, single-molecule PAINT used for the detection of individual molecules in super-resolution microscopy [117] depends on the hybridization characteristics of sequence-specific probes to their targets. It might thus benefit from additional insights into the kinetics of the hybridization of nucleic acid probes or from a combination of its approach with open-space microfluidic devices.
By overcoming some of the key limitations of the FISH assay and by presenting approaches towards the rational design of new FISH probes and procedures, microfluidic devices present powerful tools to increase the frequency of FISH being used in diagnostic laboratories. We believe that FISH can greatly benefit from microfluidics, automation, and open-space microfluidic implementations, thus making it a robust assay providing spatial genomic and transcriptomic information.