1.1. What is Recombineering?
The ability to clone and edit genetic material is an essential component of life scientists’ toolkit, allowing research in numerous fields, from molecular biology to biochemistry and cell biology to biophysics. One genome editing method known as recombineering, a portmanteau of recombination mediated genetic engineering, allows DNA manipulation without restriction enzymes or other in vitro enzymatic treatments [1]. Recombineering was initially developed for editing DNA within Escherichia coli using bacteriophage proteins, taking advantage of either the bacteriophage lambda (phage λ) Red (identified via recombination-deficient mutations) recombination system [2], [3], [4] or the Rac prophage RecET system [5]. Both systems combine an exonuclease for resecting dsDNA ends in the 5′→3′ direction, with an annealase for binding the resulting 3′-ssDNA overhang and annealing it to a homologous ssDNA molecule. Since first demonstrated in E. coli, recombineering has been successfully implemented in many other bacteria (Table 1), often using the exonuclease and annealase proteins from a host-specific bacteriophage. Recombineering is also the basis for related techniques such as multiplex automated genomic engineering (MAGE) [6] that can rapidly evolve new bacterial strains with enhanced functions.
Table 1. – A list of the organisms in which recombineering has been reported. * Denotes a gene name rather than a protein. Multiple entries may exist for bacterial species where recombineering is reported with different EATR pairs. Similar tables have been created by others [1,[15], [16], [17]]
| Bacterial Host Target | Exonuclease | Annealase | EATR Origin | Reference |
|---|---|---|---|---|
| Acinetobacter baumannii | ACINIS123_2462* | ACINIS123_2461* | A. baumanniistrain IS-123 | [18] |
| Agrobacterium tumefaciens | λExo | Redβ | Bacteriophage λ | [19] |
| Bacillus subtilis | N/A | GP35 | phage SPP1 | [20] |
| Burkholderia thailandensis | λExo | Redβ | Bacteriophage λ | [21] |
| Burkholderia pseudomallei | λExo | Redβ | Bacteriophage λ | [21] |
| Burkholderia sp. DSM 7029 | Redα7029 | Redβ7029 | DSM 7029 | [22] |
| Caulobacter crescentus | N/A | Redβ | Bacteriophage λ | [23] |
| Clostridium acetobutylicum | N/A | CPF0939* | C. perfringens | [24] |
| Collinsella stercoris | N/A | CspRecT | C. stercoris phage | [25] |
| Corynebacterium glutamicum | RecT | RecE | Rac Prophage | [26] |
| Corynebacterium glutamicum | OrfC | OrfB | L. pneumophila | [26] |
| Corynebacterium glutamicum | GP61 | GP60 | Phage Che9c of M. smegmatis | [26] |
| Escherichia coli | N/A | CspRecT | C. stercoris phage | [25] |
| Escherichia coli | λExo | Redβ | Bacteriophage λ | [4] |
| Escherichia coli | RecE | RecT | Rac Prophage | [5] |
| Klebsiella pneumonia | N/A | CspRecT | C. stercoris phage | [25] |
| Lactobacillus brevis | RecE homolog | RecT homolog | L. brevis KB290 | [27] |
| Lactobacillus casei | LCABL_13060* | LCABL_13040* | prophage PLE3 | [28] |
| Lactobacillus plantarum | lp_0642* | lp_0640* | prophage P1 | [29] |
| Lactobacillus reuteri | N/A | RecT1 | L. reuteri | [30] |
| Lactobacillus rhamnosus | N/A | LprRecT | Lactobacillus reuteri prophage | [23] |
| Lactococcus lactis | N/A | RecT1 | L. reuteri | [30] |
| Legionella pneumophila | N/A | ORF C | L. pneumophila | [31] |
| Mycoplasma pneumoniae | N/A | GP35 | phage SPP1 | [32] |
| Mycobacterium smegmatis | Gp60 | Gp61 | Phage Che9c | [33] |
| Mycobacterium tuberculosis | Gp60 | Gp61 | Phage Che9c | [34] |
| Photorhabdus luminescence | Pluα | Pluβ | P. luminescence | [16] |
| Pseudomonas aeruginosa | N/A | PapRecT | P. aeruginosaphage | [25] |
| Pseudomonas aeruginosa | λExo | Redβ | Bacteriophage λ | [35] |
| Pseudomonas putida | N/A | Rec2 | P. putida | [36] |
| Pseudomonas syringae | RecEPsy | RecTPsy | P. syringae | [37] |
| Saccharomyces cerevisiae | N/A | Redβ | Bacteriophage λ | [38] |
| Salmonella enterica | λExo | Redβ | Bacteriophage λ | [39] |
| Shigella sonnei | λExo | Redβ | Bacteriophage λ | [40] |
| Shigella flexneri | λExo | Redβ | Bacteriophage λ | [40] |
| Shigella dysenteriae | λExo | Redβ | Bacteriophage λ | [40] |
| Shewanella oneidensis | N/A | W3 Beta | Shewanella sp. W3-18-1 | [41] |
| Sinorhizobium meliloti | λExo | Redβ | Bacteriophage λ | [42] |
| Staphylococcus aureus | N/A | EF2132* | Enterococcus faecalis | [43] |
| Vibrio natriegens | SXT-Exo | SXT-Beta | SXT mobile genetic element | [44] |
| Xenorhabdus stockiae | Pluα | Pluβ | P. luminescence | [16] |
| Xenorhabdus stockiae | XBJ1_1172* | XBJ1_1171* | N/A | [45] |
| Yersinia pseudotuberculosis | λExo | Redβ | Bacteriophage λ | [46] |
| Zymomonas mobilis | RecE | RecT | Rac Prophage | [47] |
The Red and RecET phage systems have been exploited for recombineering due to their simple, streamlined, and highly efficient pathway for homologous DNA recombination known as single strand annealing (SSA). SSA is one of the three main pathways used in eukaryotic cells for the repair of dsDNA breaks, along with non-homologous end joining (NHEJ) and homologous recombination (HR) [7], [8], [9], [10], [11]. While numerous informative reviews covering different aspects of recombineering are currently available [1,[12], [13], [14]], most pay only limited attention to the structures and mechanisms of the proteins that are the key workhorses behind the method. As structural knowledge of a protein can dramatically improve our understanding of its function, this review will focus on the structures of the exonuclease and annealase proteins that have been determined to date, including the annealase structures reported during the past year. By digging deep into the structures of these proteins, we can understand not only how they function within their native bacterial hosts but also how we can continue to expand and improve recombineering in the future.
1.2. The Roles of EATR Proteins in Single-Strand Annealing
Recombineering utilizes bacteriophage proteins that catalyze homologous DNA recombination. These proteins form an Exonuclease-Annealase Two-component Recombinase system, or EATR. The term SynExo (Synaptase Exonuclease pair) has also been used, in which case synaptase is synonymous with annealase. The terms annealase, synaptase, recombinase, and single strand annealing protein (SSAP) have all been used to refer to the same group of proteins. Herein, we use the term annealase to describe proteins that bind to ssDNA and catalyze the annealing of two homologous ssDNA strands in an ATP-independent manner. Prominent examples include RecT from E. coli [48], Redβ from phage λ [49,50], Rad52 from yeast and humans [10], and ICP8 from Herpes Simplex Virus 1 (HSV1) [51,52]. Annealases typically do not catalyze DNA strand-invasion reactions (insertion of a ssDNA strand into a homologous dsDNA molecule) like the RecA and RAD51 recombinases that are ATP-dependent [53]. Instead, the two EATR proteins work in concert to catalyze DNA recombination by single strand annealing (SSA): the exonuclease binds to a dsDNA end and caries out 5ʹ→3ʹ end-resection to form a long 3′-ssDNA overhang, to which the annealase binds and anneals it to a homologous ssDNA. The two steps of the reaction, end-resection and annealing are coupled to one another via a protein-protein interaction between the exonuclease and annealase [54,55].
Before discussing the mechanisms of EATR proteins in recombineering, it is worth considering their natural roles in the propagation of the bacteriophage that typically encode them. In this regard, the Red system from phage λ has been studied in the most detail. The Red genes are not required for viability of phage λ, but they significantly enhance (by 6 to 10-fold) the number of phage particles produced upon lysis [13]. Exactly how recombination promotes phage propagation is not fully understood, but roles in replication [56], generation of concatemeric genomes for viral genome packaging [57], repair of dsDNA breaks for CRISPR evasion [58], and generation of genetic diversity [59,60] have been proposed. Although the exact mechanisms by which EATR proteins promote recombination in cells are still under investigation, several models have been proposed.
Stahl et al. described two models for phage λ recombination in E. coli, one that is dependent on the host RecA DNA strand-exchange ATPase, and another that is RecA-independent [61]. These models have been reviewed in excellent detail by Murphy [13]. Briefly, the RecA-dependent model, which is prevalent in non-replicating cells, starts with digestion of a dsDNA end by λExo, which loads Redβ onto the nascent 3′ ssDNA overhang [61]. The host RecFOR proteins then facilitate replacement of Redβ with RecA on the 3′-overhang, which promotes invasion of the 3′-overhang into a homologous dsDNA molecule, and recombination proceeds from there via the normal host double-strand break repair system [13,61].
The RecA-independent model, also known as single strand annealing (SSA), is prevalent in replicating cells. In this model, λExo and Redβ again function in concert to form a 3′-overhang bound by Redβ, which in this case directly catalyzes annealing of the 3′-overhang to a homologous ssDNA molecule. This pathway requires two DNA breaks at non-allelic sites on separate phage λ chromosomes, which, upon end-resection, can produce 3ʹ-overhangs with complementary regions that can be directly annealed to one another. Following annealing, the excess non-homologous overhanging strands are removed, and any gaps formed are filled in by a polymerase. Finally, a ligase can seal the remaining nick to result in a fully repaired functional dsDNA molecule [13,61].
A similar type of SSA pathway can ensue when a dsDNA break occurs between two directly repeated sequences on the DNA. In such a scenario, the two 3′-overhangs formed by end-resection will have homologous sequences that can be directly annealed to one another. This results in repair of the dsDNA break, but with deletion of one of the two repeats along with the sequence between them. Although the phage λ chromosome doesn't have directly repeated regions, a very similar type of SSA pathway can be highly significant in eukaryotic cells, where such repeats are common [62]. Moreover, homologs of the RecET and Red EATR proteins encoded on the IncC conjugative plasmid replicating in Salmonella enterica have been demonstrated to use this type of SSA to evade dsDNA breaks formed by a host CRISPR system [58]. Thus, the EATR-promoted SSA pathway operating at repeated DNA sequences can be relevant to bacteriophage (or plasmid) propagation, and studies of the RecET and Red proteins have served as a model for understanding double-strand break repair by SSA in humans.
The recombination model that is most relevant to recombineering is the RecA-independent SSA model that relies on Redβ annealase [63,64]. Recombineering can either employ synthetic ssDNA oligonucleotides as the input DNA electroporated into cells (typically in the range of 35-100 nucleotides), or a dsDNA cassette that can be much longer (5 - 10 kb). The dsDNA cassette is typically generated by PCR using primers containing terminal homologies to the recombination target site. In the case of oligonucleotides, the exonuclease component of the EATR is not required for recombination: the annealase binds the input oligonucleotide directly and anneals it to the target site exposed as ssDNA at the lagging strand of a replication fork [65]. By contrast, recombineering with dsDNA as the input DNA requires end-resection by the exonuclease, and the resulting 3’-ssDNA overhang is bound by the annealase. Interestingly, it appears that in dsDNA recombineering, the exonuclease typically digests one complete strand of the input dsDNA, and Redβ anneals the intact opposing strand to the target site by the same mechanism as for short oligonucleotides, at the lagging strand of a replication fork [63,64]. While recombination via the classical SSA pathway (i.e. end-to-end annealing) can occur during recombineering, the efficiency is lower due to the requirement of appropriately positioned dsDNA ends. Hence, in most cases the annealing events during recombineering occur at the replication fork.
As mentioned above, the classical RecA-independent SSA pathway in phage λ described by Stahl et al. [61] was greatly stimulated in replicating cells, although the reason for this was unclear. It was suggested that the role of replication in these experiments could be to generate appropriately positioned dsDNA ends resulting from rolling circle replication [13,61]. However, this type of SSA model did not account for the observed level of recombination in a phage λ infection, and it is conceivable that some annealing events may occur at the replication fork, as seen for recombineering [13]. Models for phage λ recombination involving replication have thus been proposed, including one involving a Replisome Invasion/Template Switch [66]. It seems unlikely that the full 50 kb phage λ chromosome would be digested during productive recombination as described above for dsDNA recombineering, but λExo is highly processive and can digest full dsDNA substrates of that length in vitro [67,68]. In any case, the use of phage EATR proteins in recombineering, as well as structural and biophysical studies of them have clearly shed new light on the possible mechanisms of phage λ recombination that have been studied for so many years by geneticists [69].
Lastly, while EATR proteins form a complex with one another, they also interact with host proteins to facilitate recombination. Most prominently, Redβ from bacteriophage λ binds to E. coli single-stranded DNA binding protein (SSB) [23,70], which coats the ssDNA at the lagging strand of the replication fork to protect it from nucleases and control access of numerous replication proteins. This interaction with SSB is absolutely required for recombination in vivo [70], presumably to displace SSB and allow Redβ to gain access to the lagging strand. Redβ from phage λ also interacts with other proteins, including phage λ replication protein P, integrase, and antitermination protein [71], although the roles of these interactions are unclear. The interactions with host proteins are even more prevalent for EATR proteins from viruses that infect eukaryotic cells. For example, ICP8, the annealase from HSV-1, interacts with other proteins in the HSV-1 replisome, such as the UL9 origin-binding protein [72] and ICP27 that is essential for the regulation of viral gene expression [73].
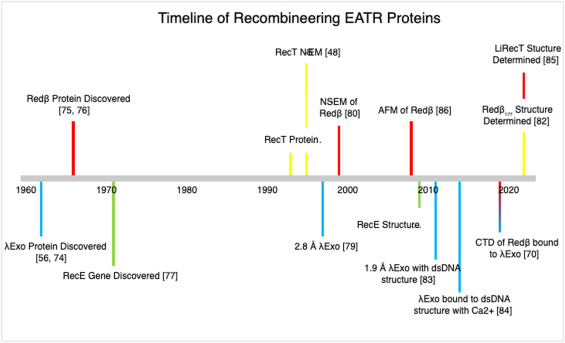
In summary, structural information on EATR proteins and their interactions with other viral and host proteins is of broad interest for understanding multiple aspects of genome maintenance including replication, repair, and generation of genetic diversity. While the RecT and Redβ annealases that have been predominantly employed for recombineering have been studied for over half a century, the key structural insights into these proteins have come relatively recently, as summarized in the historical timeline in Figure 1. The recent breakthroughs in annealase structures have put a new spotlight on these proteins and how understanding their structure can help unravel their function and lead to improvements in recombineering in the future.
 Figure 1. Timeline of the discovery and structural advances of EATR proteins used in recombineering. References for each discovery are indicated in the figure [48,56,70,[74], [75], [76], [77], [78], [79], [80], [81], [82], [83], [84], [85], [86]]. Yellow indicates discoveries for RecT, red for Redβ, blue for λExo, and green for RecE.
Figure 1. Timeline of the discovery and structural advances of EATR proteins used in recombineering. References for each discovery are indicated in the figure [48,56,70,[74], [75], [76], [77], [78], [79], [80], [81], [82], [83], [84], [85], [86]]. Yellow indicates discoveries for RecT, red for Redβ, blue for λExo, and green for RecE.2.1. Exonucleases Structures
While recombineering with single-stranded oligonucleotides as the electroporated input DNA (often referred to as single-stranded oligonucleotide repair or ssOR) only requires the annealase [65], when the input DNA is double-stranded, both the annealase and exonuclease components of a specific EATR pair are needed. In addition to recombineering, exonucleases as stand-alone enzymes have been exploited for critical roles in other biotechnology applications such as generating ssDNA from dsDNA for PCR [87], CHIP-EXO protein-DNA foot-printing [88] and generating ssDNA for several biosensor applications (a few examples include [89], [90], [91], [92]). Despite these many uses, little is known about how these proteins are evolutionarily related to one another, especially when compared to the work done for grouping annealases, discussed below [10,93].
The structures of the two main exonucleases used in E. coli recombineering, λExo from phage λ [70,79,83,84] and RecE from Rac prophage [81], have been determined by x-ray crystallography (Fig. 2). Remarkably, despite having limited sequence identity, both exonucleases form ring-shaped oligomers with central funnel-shaped channels, although λExo forms a trimer and RecE a tetramer. In both structures, the dsDNA is thought to enter at the open end of the ring, such that the 5′-strand can feed into one of the active sites to be digested into mononucleotides. The 3′-overhang then exits out the back of the channel to tether the ring to the DNA as it moves forward digesting the 5′-strand [81,83,84]. This same oligomeric architecture has been seen for λExo, RecT, and for a third member of this family whose structure has been determined, the alkaline Exonuclease from Laribacter hongkongensis [94,95].
 Figure 2. Structures of λExo and RecE. A. Surface electrostatic view of the λExo trimer (1AVQ). The wide end of the central channel to which the dsDNA substrate would enter is facing the viewer. B. Cartoon view of the λExo trimer complexed with dsDNA substrate (3SM4). The 5′-end of the orange DNA strand binds in the active site of the cyan subunit, near two Mg2+ ions. Side chains discussed in the text are labeled. C, D. Surface electrostatics and cartoon view of the RecE nuclease domain tetramer (3H4R). The view is again looking down into the wide end of the central channel. Notice that the central channels in both structures narrow at the back end.
Figure 2. Structures of λExo and RecE. A. Surface electrostatic view of the λExo trimer (1AVQ). The wide end of the central channel to which the dsDNA substrate would enter is facing the viewer. B. Cartoon view of the λExo trimer complexed with dsDNA substrate (3SM4). The 5′-end of the orange DNA strand binds in the active site of the cyan subunit, near two Mg2+ ions. Side chains discussed in the text are labeled. C, D. Surface electrostatics and cartoon view of the RecE nuclease domain tetramer (3H4R). The view is again looking down into the wide end of the central channel. Notice that the central channels in both structures narrow at the back end.2.2. λExo Structure
λExo is a highly processive alkaline exonuclease that initiates digestion at dsDNA ends. The rate of dsDNA digestion is 5-40 nucleotides per second, as determined both at the single molecule level [67,68,96] and in bulk biochemical studies [83,[97], [98], [99]]. A peculiar feature of λExo is that it requires a 5′-phosphate on the dsDNA end for active digestion [76] yet binds to dsDNA with either 5′-OH or 5′-PO4 ends with roughly equal affinity [100]. As the 5′-PO4 is five covalent bonds removed from the phosphodiester bond that is cleaved in the reaction, its impact on catalytic activity but not on binding was perplexing. A clue as to the role of the 5′-phosphate came from mutagenesis studies indicating a pivotal role for Arg-28 in enzyme processivity, and an interaction of Arg-28 with the 5′-phosphate was suggested based on modeling [101].
λExo was the first recombineering protein to have its crystal structure determined. Although first crystallized in 1985, the crystals at that time only diffracted to a 6 Å resolution [102]. It was not until 12 years later that a crystal structure was determined at 2.8 Å resolution, without DNA [79]. The structure revealed a ring-shaped homotrimer with a central channel of 30Å at one end (Fig. 2a), enough to allow dsDNA to enter, but only 15 Å at the other end, allowing only ssDNA to exit [79]. The proposed DNA binding mode nicely explained the high processivity of λExo, as the ring-shaped trimer would be physically tethered to the DNA molecule as it moves along digesting it.
Over a decade later, the structure of λExo in complex with DNA substrate was determined [83] (Fig. 2b). The crystallized complex contained a 12-bp duplex with a 5′-phosphorylated 2-nt overhang at one end (a 14-mer/12-mer), the inactive K131A variant of λExo to prevent DNA digestion, and the Mg2+ ions that are required for nuclease activity. The structure showed that the DNA is indeed bound to the central channel, but significantly tilted to place the end of the DNA with the 2-nt overhang into one of the three active sites. The two nucleotides at the 5ʹ end of the DNA are bent away from the duplex and inserted into an active site cleft, while the 3′-OH of the opposing strand is positioned to exit out the back of the trimer.
The unwinding of the DNA is mediated by apolar residues, including Leu-78 that wedge into the base pairs to separate them. The 5′-phosphate of the DNA is indeed bound at the end of the active site to Arg-28, while the scissile bond is bound to two Mg2+ ions held in place by crucial acidic active site residues. The structure, which visualizes the nucleophilic water molecule that is poised for attack [83] supports a classic two-metal nuclease mechanism [103]characteristic of the type 2 restriction endonuclease (T2RE) family [104], also known as the PD-(D/E)XK family [105]. Three loops of λExo, one from each subunit, extend from the rim of the central channel to contact the downstream portion of the dsDNA substrate. The Arg-45 side chain from one of the three loops inserts into the minor groove of the DNA and is proposed to help the enzyme keep on track. In support of this role, mutation of Arg-45 to Ala almost completely disrupts cleavage activity [83,106].
Based on this structure, an “electrostatic ratchet” model for processive digestion was proposed in which the interaction of the 5′-phosphate on the DNA with Arg-28 at the end of the active site is key to moving the enzyme forward. As each mononucleotide is cleaved from the 5′-end and released with Mg2+ out the rear portal on the trimer, the newly generated 5′-phosphate of the next nucleotide on the DNA would be attracted to the positively charged pocket containing Arg-28. The hydrophobic wedge formed by Leu-78 is proposed to help unwind the base pairs as the enzyme moves along the DNA, and the Arg-45 side chain is thought to act as a rudder to help the trimer track along the minor groove of the downstream portion of the DNA.
The most recent crystal structure of λExo shows a trimer bound to three copies of the Redβ CTD, resolved to 2.3 Å [70] (Fig. 3). This structure provided the first direct insights into the architecture of the λExo-Redβ EATR complex and is remarkably consistent with a model in which the role of the interaction is to load the Redβ annealase directly onto the 3′-overhang that is formed by λExo during digestion [13,107]. Further details of this interaction will be examined below in section 2.5.
 Figure 3. Structure of λExo in complex with the Redβ CTD. A. view from the back of the λExo trimer (6M9K). The dsDNA substrate would enter the ring from the back and the 3′-overhang ssDNA would come out towards the viewer. The three Redβ CTDs are colored magenta. B. Side view showing that the three Redβ NTDs and linkers (residues 1-193) would lie on the face of the λExo trimer from which the 3′-overhang ssDNA is extruded. The structure thus suggests a mechanism in which Redβ monomers are directly loaded onto the 3′-overhang as it is formed by λExo.
Figure 3. Structure of λExo in complex with the Redβ CTD. A. view from the back of the λExo trimer (6M9K). The dsDNA substrate would enter the ring from the back and the 3′-overhang ssDNA would come out towards the viewer. The three Redβ CTDs are colored magenta. B. Side view showing that the three Redβ NTDs and linkers (residues 1-193) would lie on the face of the λExo trimer from which the 3′-overhang ssDNA is extruded. The structure thus suggests a mechanism in which Redβ monomers are directly loaded onto the 3′-overhang as it is formed by λExo.2.3. RecE Structure
While λExo is a 226 amino acid protein, RecE is a much larger 866 amino acid protein that contains a C-terminal nuclease domain (residues 564-866) and an N-terminal domain of unknown function. The nuclease domain can substitute genetically for the full-length protein [108], although full-length RecE has enhanced activity for recombineering involving linear-linear (end-to-end) SSA recombination in vivo [109]. The crystal structure of the RecE nuclease domain was determined at 2.8 Å in the absence of DNA in 2009 [81], between the time the two λExo structures without and with DNA were published. The RecE fold has a core topology similar to λExo and a common set of conserved active site residues. Intriguingly, the RecE monomers pack into the tetramer in essentially opposite orientations as λExo, relative to the end of the channel at which the DNA would enter. This suggests that although RecE and λExo are evolutionarily related at the tertiary structure level, their similar quaternary structures (RecE tetramer and λExo trimer) likely evolved independently from a monomeric ancestor. Clearly, a ring-shaped structure with a tapered central channel is a fundamental architectural feature for this processive 5′-3′ exonuclease enzyme family.
Each subunit of RecE has a channel that contains an active site connecting with a positively charged portal that could allow for the release of mononucleotides as they are cleaved (Fig. 2c). The structure was determined without DNA in the presence of Ca2+, which supports DNA binding but not cleavage. Although only one Ca2+ ion is bound per active site, two Mg2+ ions are presumably needed for cleavage. The set of critical active site residues is primarily conserved between RecE and λExo, with one notable exception: Glu-85 of λExo is replaced by His-652 in RecE. This residue is also histidine in the C-terminal nuclease domain of RecB of the E. coli RecBCD complex, another member of the T2RE family. The role of this residue in catalysis is not yet clear, but it could help to stabilize the 3’-OH leaving the group after hydrolysis.
Another difference between RecE and λExo is that RecE contains much longer loops projecting out from the rim of the central channel, presumably to capture the dsDNA substrate. These loops, formed by residues 665-698 of RecE, are largely disordered in the crystal structure and are not part of the final refined model. One of our laboratories (Bell) successfully crystallized the RecE nuclease domain in a complex with different lengths of DNA. However, the DNA could never be visualized, presumably because it did not sit down in a unique orientation relative to the crystal packing interactions. The loops did, however, become partially visualized in these structures.
In summary, the RecE and λExo structures show several common features that appear fundamental to the processive nuclease activity required for 5′→3′ end-resection. These features also appear to be conserved for the additional structures of related exonuclease proteins of the phage recombination systems that have been determined.
2.4. Evolutionary Analysis of Recombineering Exonucleases by Sequence Alignments
To gain further insights into exonuclease function, we analyzed sequence conservation in both λExo and RecE using the 2000 hit blast search results against the UniProt ref90 database. Following multiple sequence alignment and quality control of these datasets, the final MSAs consisted of 1347 sequences for λExo and 183 for RecE. Many of the RecE sequences were eliminated once sequences with greater than 90% similarity were clustered. Both λExo and RecE belong to the PD-(D/E)XK phosphodiesterase superfamily, a highly diverse group of proteins with homologs present in all domains of life [104,105]. The superfamily consists primarily of nucleases, including processive exonucleases such as λExo, RecE, E. coli RecB, and the herpesvirus alkaline nuclease UL12, as well as many restriction endonucleases including those used in traditional cloning techniques. As with most members of this superfamily, λΕxo and RecE have a conserved core fold consisting of a four-stranded, mixed β-sheet flanked by α-helices, with αβββαβ topology [105]. Embedded within this fold are the conserved aspartate, glutamate, and lysine residues that give the PD-(D/E)XK family its name.
In our analysis, the active site residues of both proteins appear to be highly conserved, and in most cases identical across all constituent sequences. In contrast, other regions of the protein are more variable (Fig. 4a & b, Supplementary Figures 1 & 2). Notably, λExo and RecE differ in the composition of their active site residues. λExo displays a highly conserved PD-EXK active site structure (Fig. 4c), whereas RecE has the alternate structure PD-DXK. In both alignments, conserved positively charged residues flank the active site and are thought to facilitate binding to the DNA substrate.
 Figure 4. Sequence conservation of λExo and RecE: Sequence conservation of a) λExo (PDBID:1AVQ) 1347 sequences, and b) RecE (PDBID:3H4R) 183 sequences. Scale values represent the entropy-based AL2CO conservation index, where blue and red correspond to the lowest and highest sequence conservation, respectively. Active site residues are indicated in black stick representation. Representative MSA is shown for c) λExo and d) RecE that summarise the full 2000 hit analysis. Secondary structural features, mapped from atomic structures of the reference sequences, are indicated above the MSA, with α-helices in orange and β-sheets in green. Active site residues are indicated above the MSA by a black diamond, and the active site region is bound by a black box. The consensus active site for each family is provided at the end of each respective MSA.
Figure 4. Sequence conservation of λExo and RecE: Sequence conservation of a) λExo (PDBID:1AVQ) 1347 sequences, and b) RecE (PDBID:3H4R) 183 sequences. Scale values represent the entropy-based AL2CO conservation index, where blue and red correspond to the lowest and highest sequence conservation, respectively. Active site residues are indicated in black stick representation. Representative MSA is shown for c) λExo and d) RecE that summarise the full 2000 hit analysis. Secondary structural features, mapped from atomic structures of the reference sequences, are indicated above the MSA, with α-helices in orange and β-sheets in green. Active site residues are indicated above the MSA by a black diamond, and the active site region is bound by a black box. The consensus active site for each family is provided at the end of each respective MSA.Of particular interest is the great disparity in length between the λExo and RecE families. While the PD-(D/E)XK-like domain spans the entire length of the λExo sequences, it comprises only the C-terminal segment of RecE. A review of the hits retrieved by RecE reveals a heterogeneity in sequence length, with some showing homology across the entire length of RecE including its large N-terminal domain. In contrast, others like λExo consist of only a single exonuclease domain (Supplementary Figure 1). As expected, the C-terminal PD-(D/E)XK domain showed the greatest conservation across all alignment regions prior to truncation, possibly explaining the higher number of sequences eliminated during the clustering step.
A review of the hits retrieved by BLAST search of λExo identified many sequences identified as homologs of YqaJ, a domain from a known EATR pair found in the skin element of Bacillus subtilis [110]. Other homologs of YqaJ include Chu exonuclease of the B. subtilis phage SPP1, which forms an EATR pair with its partner annealase GP35 [20,110].
Also of note, the elements that encode λExo and RecE have different reproductive methods. λExo is encoded within a phage that can undergo lytic reproduction, whereas RecE is present in a defective prophage replicating with the host. This difference in reproduction method could have a marked effect on sequence evolution. Phage λ has a generation time of ∼7.7 phages min−1 [111], which is >20x faster than that of E. coli, which is estimated at ∼0.3 bacterium min−1 [112]. The more rapid evolution of λExo could account for the higher level of sequence similarity for the RecE family in our analysis. Alternatively, the differences in similarity could reflect the limited number of RecE sequences available in the current UniProt Database.
2.5. The Lambda Phage EATR Complex: λExo+Redβ
The interaction between the two phage EATR proteins has been known for over half a century. In fact, the Redβ protein was discovered during the purification of λExo, as the two proteins were seen to co-purify with an apparent 1:1 stoichiometry [54]. While the nuclease activity of λExo had been well known [75,76], the function of Redβ was not established until nearly a decade later when it was discovered that Redβ could promote the annealing of homologous ssDNA strands [49]. Although Redβ can function independently, somewhat higher annealing activity was observed in the presence of λExo [49]. The reason for this is still not apparent. There is actually a third protein in the Red system known as γ-protein (also referred to as Gam), encoded by the gam gene [113], [114], [115]. The γ-protein is often not present in genomes encoding the typical EATR pair, including in the E. coli Rac prophage encoding RecE and RecT, and it appears to take on a more supplementary role. In phage λ the gam gene is required to transition from the early to the late stage of viral infection [116], but the γ-protein does not appear to interact with any other phage λ recombination proteins [117]. Instead, it binds to the host RecBCD helicase/exonuclease complex to prevent it from digesting dsDNA ends [113], [114], [115], which are present on the linear form of the λ phage genome [113]. The γ-protein can efficiently inhibit both nuclease activities of RecBCD, including its exonuclease activity on dsDNA and its ssDNA and endonuclease activity on ssDNA [114,115]. Crystal structures of γ-protein reveal a small alpha-helical dimer, and a cryo-EM structure of γ-protein in complex with RecBCD has been determined [118,119]. Red-mediated recombination with linear dsDNA can occur without γ-protein in vivo [15]. Still, γ-protein is typically included in recombineering strains with active RecBCD to prevent the destruction of linear duplex DNA.
While there is currently no structure of a complete EATR complex, there have been attempts to model what the λ phage EATR complex could look like (Fig. 5). One of the first models, proposed by Tolun in 2007, considered the available biochemical and stoichiometric data [107] and was comprised of four λExo trimers bound to a dodecameric ring of Redβ (Fig. 5a). This complex would presumably load onto a dsDNA end through one of the λExo trimers. According to the model, as the 5′-strand is digested, the exposed ssDNA would be bound to the N-terminal domains of the associated Redβ subunits [13,107]. Although the purified EATR complex has a 1:1 stoichiometry [54], this model would suggest a higher concentration of Redβ compared to λExo. Mechanistically, λExo should only need to be stoichiometric with dsDNA ends, whereas higher levels of Redβ would be required to form the larger oligomeric complexes on DNA. Presumably, Redβ monomers detach from the EATR complex to form an oligomeric complex with the nascent 3’-overhang ssDNA for recombination [82]. Indeed, expression of Redβ at higher levels than λExo leads to a significant improvement of recombination efficiency, whereas an excess of λExo over Redβ decreases recombination levels [55]. A similar relationship was observed for RecE and RecT, suggesting that the two EATR pairs work by similar mechanisms [55]. Despite this trend, when Redβ and λExo are expressed for recombineering from their natural PL promoter on a pSIM5 vector, Western blot analysis with a polyclonal antibody raised to both proteins indicates that similar levels of each are expressed [70,125]. Whether this reflects their levels at the end of a phage λ infection when recombination is most active is uncertain.
 Figure 5. Proposed models of the EATR complex of Redβ and λExo. (A) is an early model, reproduced with author permission from Tolun [107], showing four λExo trimers (yellow) bound to the C-terminal domains of Redβ (lime) that form a larger ring with their N-terminal domains (darker green). (B) is a reproduction with author permission of the model proposed by Newing et al. [82] showing a composite structural model of the complex, with Redβ highlighted in pink and λExo in blue. AlphaFold 2 was used to predict the structure of Redβ and λExo (C), showing a λExo trimer (blue) bound to three Redβ monomers (red). (D) highlights the interaction of single λExo and Redβ monomers with the experimentally determined structures of λExo (PDB ID = 6M9K) in dark grey and of the C-terminal domain (PDB ID= 6M9K) and the first 177 amino acids (PDB ID = 7UJL) in light grey.
Figure 5. Proposed models of the EATR complex of Redβ and λExo. (A) is an early model, reproduced with author permission from Tolun [107], showing four λExo trimers (yellow) bound to the C-terminal domains of Redβ (lime) that form a larger ring with their N-terminal domains (darker green). (B) is a reproduction with author permission of the model proposed by Newing et al. [82] showing a composite structural model of the complex, with Redβ highlighted in pink and λExo in blue. AlphaFold 2 was used to predict the structure of Redβ and λExo (C), showing a λExo trimer (blue) bound to three Redβ monomers (red). (D) highlights the interaction of single λExo and Redβ monomers with the experimentally determined structures of λExo (PDB ID = 6M9K) in dark grey and of the C-terminal domain (PDB ID= 6M9K) and the first 177 amino acids (PDB ID = 7UJL) in light grey.While the first EATR model was primarily based on biochemical data, Newing et al. [82] proposed a model based on newly available structural data (Fig. 5b). This model incorporated the structures of λExo bound to DNA [83] and to the CTD of Redβ [70], combined with the Redβ177 cryo-EM structure [82]. AlphaFold 2 was used to predict a structure for the linker region of Redβ (residues 178-193), which has not yet been resolved experimentally. This model also assumed higher levels of Redβ than λExo as the long 3′-ssDNA overhang generated by λExo trimer digestion would require multiple Redβ monomers to form a continuous protein-DNA filament seen in the structure.
Here we propose a third possible model for the phage λ EATR complex, generated using AlphaFold 2 (Fig. 5c,d). The model contains three λExo and three Redβ subunits, and retains the signature 1:1 ratio, with λExo forming the characteristic trimer, three Redβ N-terminal domains interacting with one another as in the Redβ177 structure, and three Redβ C-terminal domains bound to λExo as in the crystal structure of the complex [70]. While this model is asymmetric in how the N-terminal domains are positioned, the Redβ linker region is likely to be flexible enough to allow for the conformational differences. Interestingly, the cleft on the Redβ N-terminal domain that contains DNA in the cryo-EM structure is occupied by a new α-helix from the linker region that is generated by AlphaFold 2 [82]. This α-helix would block DNA binding and could conceivably control how Redβ monomers assemble on the nascent ssDNA that is generated by λExo. While there is still no experimentally determined structure of the full phage λ EATR complex, the insights we gain from each new structure and model help to assemble the pieces.
3.1. Annealase Proteins
There are many distinct types of proteins with annealase activity found in nature, as first mapped out by Iyer et al. who proposed three distinct superfamilies grouped around ERF (essential recombination function), RecT/Redβ, and Rad52. Each family was predicted to have a different core fold and a distinct pattern of sequence conservation [10]. Lopes et al. later proposed a different grouping based on Rad52-like, Gp2.5-like, and Rad51-like sequences [93]. Most recently, seven annealase families were proposed including Sak3, Sak4, Rad52/22, ERF, RecT/Redβ, Gp2.5, and RecA [120]. The latter two groupings included Rad51/RecA family proteins that have annealase activity, but primarily function in ATP-dependent DNA strand exchange for homologous recombination [53]. Similarly, Gp2.5 is a single-stranded DNA binding protein from bacteriophage T7 that presumably has annealase activity as a side effect of ssDNA-binding [121]. Due in part to the diversity of annealase proteins, we have yet to arrive at a consensus mechanism for how they catalyze DNA annealing. Based on their distinct core folds and presumably different evolutionary origins, the different types of annealase proteins could indeed operate by different mechanisms. Of the 7 families described most recently, only three have representative high-resolution structures available, namely the Rad52, RecA/RAD51, and RecT/Redβ families.
Recombineering has primarily been developed and optimized for use in E. coli, and the RecET and phage λ Red proteins have evolved to function in E. coli. The annealase activity is now known to depend on an interaction with the host SSB protein [23,70], which will vary in sequence in different hosts. Therefore, it can be challenging to predict if λ Red or RecET will be functional in a given bacteria of interest. However, recombineering can be expanded to new organisms by mining for EATR proteins from a bacteriophage (or prophage) that infect them (Table 1). Moreover, Redβ from phage λ functions efficiently as an annealase for recombineering in close relatives of E. coli including Salmonella enterica [39]. The interaction between annealases and host SSB proteins largely involves the last ∼9 residues of SSB [23,70], which is the site for interaction of numerous E. coli host replication proteins [122]. Altering this sequence has allowed the portability of a given annealase into a new bacterium of interest [23], providing a simple means to increase the efficiency of recombineering in new bacterial hosts. Knowledge of how RecT and Redβ operate can also benefit our understanding of the annealase mechanism, for which there has been a general lack of structural information, particularly for the relevant protein-DNA complexes. While structures with ssDNA substrate have been available for eukaryotic annealases including Rad52 and ICP8, the recent structures of RecT and Redβ [82,85] have been determined in complex with a duplex intermediate of annealing, and therefore provide important new insights into the possible annealing mechanism, as will now be described.
3.2. Structure of Redβ
While Redβ was discovered over half a century ago [75,76], structural investigations only began approximately 20 years ago. The first structures reported by Passy et al. used negative staining electron microscopy (NS-EM) [80] and revealed oligomeric rings in the absence of DNA and larger rings with ssDNA. Left-handed helical filaments were observed when Redβ was mixed with heat-denatured double stranded DNA, which was the first indication of a structural transition upon annealing [80]. Almost ten years after Passy et al.’s findings, further investigations using atomic force microscopy (AFM) [86]revealed similar helical filaments in the presence of two complementary ssDNA sequences, but disperse monomers bound to a single ssDNA sequence. A model for annealing was proposed in which a clamped dimer of Redβ stabilizes a nucleus of complementarity from which annealing can propagate [86].
While the work of Erler et al. confirmed that Redβ forms ring-like structures in the absence of DNA, it more clearly showed structures resembling a split-lock washer, with a gap or a slight overlap between monomers at one end of each ring [86]. Unlike NS-EM, which gives 2D projection, AFM imaging is sensitive to height, which explains how split-lock washers were detected. As there were some differences in the oligomeric complexes seen by different low resolution imaging methods, a high-resolution structure of Redβ was clearly needed to resolve the discrepancies.
The first atomic structure of Redβ was of its C-terminal domain, determined in complex with a λExo trimer. The overall architecture of the complex supported a model in which Redβ is loaded onto ssDNA during DNA end-resection by λExo [70]. From a mutational analysis of the λExo-CTD interface, a second role for the CTD in binding to the host SSB protein was discovered. The two interactions were found to use an overlapping site and are thus likely to be mutually exclusive. A ‘hand-off’ model was proposed in which the interaction with λExo loads Redβ onto the first ssDNA (the 3’-overhang formed by λExo). In contrast, the subsequent interaction with SSB localizes the initial Redβ-ssDNA complex to the lagging strand of the replication fork, where it can scan the lagging strand for a sequence that is complementary to the first ssDNA [65,70]. The structure of the Redβ CTD is also significant because the other available structure of Redβ that would become available only includes its N-terminal DNA-binding domain [82].
Most recently, cryo-EM revealed the structure of the Redβ N-terminal domain (NTD) that is responsible for DNA binding and oligomerization (Fig. 6b). Rather dramatically, the structure captured a helical filament of Redβ in complex with a novel intermediate of DNA annealing that has an unusual conformation of duplex DNA [82]. The structure used a truncated form of Redβ that only included its first 177 amino acids (out of 261 in native Redβ). The cryo-EM 2D class averages showed 1- and 2- start helical filaments, with the start of a helix denoting the number of threads that are found per turn of a helix [82]. While 2-start filaments have also been observed for the ICP8 annealase from HSV-1, their functional role is not clear [124,138]. The 1-start filaments of Redβ177 on the other hand suggested a compelling mechanism for annealing.