1. Introduction
Tissue engineering has emerged over the last decades due to a high need to repair or replace diseased and injured tissues and organs. In tissue engineering, cells are combined with growth factors and scaffolds which provide physical support for the cells and aid with the formation of a three-dimensional (3D) tissue [1]. Conventionally, fabrication methods such as particle leaching [2], gas foaming [3] or electrospinning [4] are used to create scaffolds for tissue engineering. However, these aforementioned scaffold fabrication techniques do not offer precise control over the porosity, pore interconnectivity or spatial distribution of the pores. 3D printing has recently attracted much attention due to the fact that it has the potential to overcome the shortages of traditionally employed scaffold fabrication technologies [5]. In extrusion-based 3D printing, scaffolds are created by deposition of materials in a layer-by-layer fashion to create 3D structures. The major advantages of 3D printing are 1) the easily customizable external shape, 2) the high control over the internal architecture of the scaffold, and 3) the automated process to achieve consistent results. This allows the creation of scaffolds with a pre-determined pore size, porosity and pore interconnectivity, which is especially important in the areas of bone tissue engineering, where the pore size is a critical parameter that determines vascularization and osteointegration of a bone scaffold [6].
The development of inks for the extrusion based 3D printing of biomaterials (a type of biofabrication [7]) is one of the grand challenges to propel the technology forward. With a larger variety of biomaterial inks available, extrusion based biofabrication would become more attractive to a wider audience of researchers which might help to drive this technology towards clinical applications. Besides assessing the printability of newly created inks as outlined recently by Paxton et al. [8], great care should be taken throughout the entire development process regarding scalability and constant quality control of the manufactured inks [9]. It is therefore crucial to work with robust polymer synthesis methods and to use only materials and technologies which allow the manufacturing of large batches of ink. Here we describe the development and characterization of a potential new ink for the 3D printing of a bone layer of biphasic constructs such as osteochondral grafts. The ink is based on gellan gum methacrylate (GG-MA), hyaluronic acid methacrylate (HAMA) and hydroxyapatite (HAp) particles. Gellan gum (GG) based inks have recently been shown to have excellent printability [10] and both hyaluronic acid [11,12] and hydroxyapatite [[13], [14], [15]] have been used in the past for bone tissue engineering applications. HAp can also be used to modify the rheological behaviour [16,17] of GG-MA to enhance its printing properties. Methacrylation of the polymer components of the ink was chosen to facilitate crosslinking between two different materials (phases) of a biphasic constructs as well as to enable the creation of a strong interface between layers within the same phase.
Being of biological origin, gellan gum (as many other biopolymers) suffers from batch-to-batch variations and chemical modification of a particular batch of gellan gum can exacerbate this issue even further. To achieve consistent printing results, robustness of the synthetic procedure for gellan gum methacrylate is therefore crucial. Batch-to-batch variations in the degree of methacrylation influence not only the final mechanical properties of the printed graft but might as well influence the rheological properties of the ink [18]. In inks such as gelatine methacrylate (GelMa), methacrylate groups are introduced using methacrylic anhydride through the modification of the amine moieties present in the chain [19]. In other materials, such as hyaluronic acid, the methacrylic anhydride targets the primary alcohols [20]. Another method for methacrylation, which can target both carboxylic acids and primary alcohols [21], is the usage of glycidyl methacrylate (GMA) [[22], [23], [24]]. For this method, a stable pH of 8.5 during the reaction is needed in order to achieve high degrees of substitution within 24 h. At pH 8.5, the transesterification reaction of GMA with the carboxylic acid moiety takes place, which proceeds faster and is therefore preferred over the slower ring-opening reaction on the epoxy group [25]. In the case of biopolymers such as gellan gum, a too high or too low pH further increases the risk of depolymerisation of the biopolymer chain [26]. Manual or automated adjustment of the pH to 8.5 has major drawbacks. Manual pH adjustment can lead to inconsistent degrees of methacrylation due to an unstable pH over time. Automated titrators negate the need for manual pH adjustments but measure the pH at a fixed position in the reaction vessel. This becomes problematic with larger reaction volumes for large lab scale synthesis, as the measured pH might no longer be representative of the entire vessel which causes the titrator to add the wrong amounts of acid or base into the reaction. Further, in the case of biopolymers such as gellan gum, the reactions are preferably carried out at low polymer concentrations to avoid high solution viscosities which would cause a reduction in mass transfer as well as an inconsistent pH throughout the reaction vessel.
The first goal of the presented work was therefore to develop a new method for the synthesis of large batches (5 g) of GG-MA using buffered solutions to control the degree of methacrylation. GG-MA synthesis is commonly carried out on the 1 g scale [27,28] and occasionally on a 3 g scale [29], however, bigger batch sizes would enable the fabrication of larger ink volumes (in our case >100 ml) from a single batch of GG-MA, therefore reducing the potential for inconsistent printing results using cartridges from different ink production dates. The second goal of this work was to develop and evaluate methods to assess the reproducibility of the printing process. To assess how needle diameters affect the printing consistency, we compared different needle geometries and ink compositions through extrusion force measurements. These measurements can also be used to evaluate ink homogeneity within the cartridge and therefore the potential to achieve consistent printing results. Similar setups have been used in the past to assess the filter pressing effect and evaluate the injectability of bone cement pastes [30], but has to our knowledge not often been used to assess inks for 3D printing. Extrusion force measurements can not only show if air bubbles were introduced during the cartridge filling, but also reveal potential clogging due to aggregates or larger particles present in the ink. Particles such as hydroxyapatite [31] or laponite [32] are often added to inks to introduce biologically relevant materials or simply as rheological modifiers. Extrusion force measurements therefore enable us to choose the right needle size and shape for such particle containing inks to avoid clogging. Both air bubbles and clogging can lead to inconsistent printing results due to the altered flow during extrusion of the ink and are, although not often mentioned, common obstacles during scaffold fabrication using 3D printing and can jeopardize the mechanical integrity of larger scaffolds. Inks with a large content of solid particles are also not only prone to clogging but also to inhomogeneity within a cartridge due to insufficient mixing, particle aggregation or, in the case of lower viscosity inks, particle sedimentation. To further assess the ink homogeneity throughout the different locations of a printer cartridge, the ink compositions from different sections of the cartridge were evaluated with thermogravimetric analysis (TGA). To complete the evaluation of the printed constructs, the interfacial bond strength between two printed layers was analyzed with the help of push-out tests. Finally, we investigated the viability and ability of mesenchymal stromal cell (MSCs) to adhere to the inks and analyzed the surfaces of different ink compositions using scanning electron microscopy (SEM).
2. Materials and methods
2.1. Materials
All materials and reagents were purchased from Sigma-Aldrich, Invitrogen, Thermo Fisher Scientific or Merck Millipore and were used as received unless otherwise stated. The HAp particles were obtained from Acros Organics.
2.2. Methods
2.2.1. GG-MA synthesis
GG-MA was synthesized in two ways: with manual pH control as previously described [23] or by using a buffered solution. For the synthesis with manual pH control, 1 g of low-acyl gellan gum (Kelco, USA) was dissolved in 100 ml of Milli-Q water and heated to 90 °C in a sealed Erlenmeyer flask. After the solution became transparent, indicating complete dissolution of the gellan gum, it was cooled down to room temperature under vigorous stirring. The pH was adjusted to 8.5 with 0.5 M NaOH and a 20-fold molar excess of glycidylmethacrylate (GMA) was added. The pH of the reaction was manually maintained for 3 h through drop wise addition of 0.1 M NaOH. The reaction was allowed to proceed for a total of 24 h after which the solution was precipitated in 50 ml of cold acetone. The gel-like precipitate was placed in dialysis bags (12–14 kDa MWCO) and dialyzed against Milli-Q water for 7 days with water changes every hour during the first 6 h and twice daily for the remaining 6 days. The product was finally freeze-dried and stored at −20 °C.
For the buffer-mediated synthesis, gellan gum was dissolved in 2-amino-2-methyl-1,3-propanediol (AMPD) buffer (100 mM, pH 8.5) at 1% w/v for batch sizes of up to 5 g. GG was dissolved in the buffer in a sealed flask at room temperature and then heated up to 90 °C under vigorous stirring until a clear solution was obtained. The solution was then cooled down to room temperature under vigorous stirring and GMA was added to the solution at various molar excesses ranging from 2.5 to 20 equivalents with respect to the carboxylic acid present in gellan gum. After 24 h, the polymer was precipitated and purified in the same way as when synthesized with manual pH control. The degree of substitution (DS) was calculated by 1H NMR (D2O, 500 MHz, 80 °C) using the ratio of the integrals of the peaks from the methacrylate groups at 5.9 ppm and 6.3 ppm and the peak of the methyl group belonging to the rhamnose unit of gellan gum at 1.45 ppm. The structures of both GG and GG-MA are shown in Fig. S1.
2.2.2. HAMA synthesis
HAMA was synthesized as described previously in the literature [33]. Briefly, 1% low molecular weight HA (280 kDa) was dissolved in ultrapure water overnight. The next day, the solution was placed in an ice bath and the pH was adjusted to 8.0 with NaOH. A 20-fold molar excess (per disaccharide unit) of methacrylic anhydride was then added under vigorous stirring. During the first 3 h, the pH was maintained at pH 8.0 by adding 5 M NaOH after which the reaction was allowed to proceed for 24 h at 4 °C. Unlike GMA, methacrylic anhydride does not react via two different reaction mechanisms depending on the pH which is why a constant pH over 24 h is less critical. After 24 h, HAMA was precipitated by dripping the solution into an excess of ice-cold ethanol. The precipitate was then collected by vacuum filtration. After redissolving the precipitated HAMA in ultrapure water, it was dialyzed for 3 days against ultrapure water (Spectra/Por 5, MWCO 12–14 kDa). The water was changed every 24 h. The product was freeze-dried and stored at −20 °C before use. NMR spectroscopy (D2O, 500 MHz) revealed successful methacrylation of the hyaluronan. The degree of substitution (DS) was calculated by the ratio of the integrals of the vinyl protons at 5.6 ppm and 6.1 ppm to the protons from the methyl groups of both, HA and the methacryloyl residues at 1.9 ppm. The calculated degree of substitution for the batches utilized in this work ranged from 31.2–34.6%.
2.2.3. Hydroxyapatite particle analysis
The size distribution of the HAp particles was evaluated by means of light scattering using a Malvern MasterSizer 2000. The particles were suspended in degassed water using a sonicator probe and analyzed immediately to avoid particle sedimentation. The size distribution of the HAp particles is shown in Fig. S2.
2.2.4. Size exclusion chromatography
Size exclusion chromatography (SEC) was performed to determine the influence of the synthesis and cryomilling on the molecular weight of GG and HA and their derivatives. Each sample was solubilized in 100 mM NaNO3 containing 5 mM of the eluent NaN3. Solubilization took place over night at 37 °C for the HA samples and at 70 °C for the GG sample to prepare 0.1% solutions for analysis. Before injection, the samples were passed through a 0.45 μm filter. The HPLC setup used for the SEC measurements included a binary pump, degasser, thermostated column compartment and an auto sampler, all from HP (Series 1100, Hewlett Packard, USA). A pre-column (Viscotek AGuard Col. 50 × 6.0 mm, Malvern Instruments Ltd., United Kingdom) was used together with a A5000 column (Viscotek, 300 × 7.8 mm, Malvern Instruments Ltd., UK) and a suprema 30,000 column (10 μm, 8 × 300 mm, PSS Polymer Standards Service GmbH, Germany). The temperature of the columns was kept at 35 °C with a flow rate of 1 ml/min and an injection volume of 50 μL. The elution was recorded using a refractive index detector (Series 1200, Agilent Technologies, Switzerland). The average molecular weights were calculated based on the measured standard curve with the ChemStation software (ChemStation for LC 3D systems, Rev. B.04.02 SP1) and the add-on Cirrus GPC/SEC software (version 3.4.1) from Agilent.
2.2.5. Ink preparation and cartridge loading
To facilitate dissolution of the lyophilized polymers (retrieved as white sponges), GG-MA and HAMA were milled using a cryomill (Retsch, Switzerland, 25 ml grinding jar, 15 mm Ø grinding balls). GG-MA and HAMA were placed into a pre-cooled grinding jar at a weight ratio of 2.67:1 and milled for 2 min at −196 °C at a frequency of 30 Hz. A fine powder was obtained which was stored at −20 °C until further use. For the mixing of the materials, a planetary centrifugal mixer (ARE-250 CE, ThinkyMixer, Japan) was utilized. Milli-Q water was placed in a container and GG-MA/HAMA powder was added to achieve a final concentration of 4% w/v GG-MA and 1.5% w/v HAMA (from here on named “ink”). Different amounts (% w/w) of hydroxyapatite particles were then added to the container and mixed for 20 min at 2000 rpm. A warm paste was obtained from the mixing process which was further stirred with a spatula until the paste cooled down to room temperature. The ink was stored in the mixing container at 4 °C until utilized for printing. Before loading the ink into the printer cartridge, the photoinitiator Irgacure 2959 (Ciba, Switzerland) was added to obtain a final concentration of 0.05% (w/v). The ink containing Irgacure 2959 was filled into the printing cartridges (3 cc, Nordson EFD, Switzerland) using an Omnifix syringe (Braun Medical Inc., USA) and a female luer-lock connector piece. Care was taken not to trap air during the loading procedure. In case air was trapped, centrifugation of the loaded cartridges was performed at 500 rpm (Z400K, Hermle, Germany).
2.2.6. Rheological measurements
The influence of the GG-MA synthesis procedure, hydroxyapatite content as well as polymer processing on the rheological properties of the ink was assessed using an MCR 301 rheometer (Anton Paar, Switzerland). The rheometer was equipped with a Peltier element, a thermostatic hood for temperature control and a UV Source (Omnicure Series 1000, 320–500 nm wavelength, 9.55 mW/cm2) (Lumen Dynamics, Canada) for UV-crosslinking. The samples were covered with a low viscosity silicon oil to avoid evaporation of water from the samples and all measurements were performed at 25 °C. A profiled parallel-plate (20 mm diameter, 0.5 mm profile) setup was used to avoid wall-slip. The flow properties of the inks were evaluated through a shear rate sweep from 0.001 to 1000 s−1. The shear recovery experiments were performed under oscillation by measuring the storage (G′) and the loss modulus (G″) at an angular frequency of 5 rad/s and a low strain of 0.01% which was found to be within the linear viscoelastic range (LVR). The strain was then increased to 150% at the same frequency for 150 s, before returning back to 0.01% strain for 600 s. For the UV-crosslinking of the ink, G′ and G″ were recorded for 60 s before illuminating the samples with UV-light for 300 s while measuring the evolution of G′ and G″. To assess the relaxation behaviour of the ink, the normal force upon compression of the sample down to a 0.5 mm gap was recorded over the course of 60 min. All rheological curves represent the average of two measurements of separate samples.
2.2.7. Extrusion force measurements
Extrusion force measurements were performed on a Texture Analyzer XT Plus (Stable Micro Systems, United Kingdom) equipped with a custom-made extrusion setup (link to the CAD files can be found in the supplementary information). Printer cartridges were filled with inks containing different amounts of HAp and placed into the holder. Total ink volume in the cartridges differed between the different samples. The plunger of the cartridges was pushed down at a constant displacement rate of 0.25 mm/s until all ink was pushed out of the cartridge. The force was assessed using a load cell (max. load 5 kg, Stable Micro Systems).
2.2.8. Thermogravimetric analysis
TGA measurements were performed to assess the hydroxyapatite content in the ink from different parts of a 3 cc printer cartridge which was prepared according to Section 2.2.5. Samples were measured on a TGA/SDTA851e (Mettler Toledo, Switzerland) in air. 3 cc printer cartridges were filled with ink and extruded using the printer at a pressure 95 kPa. Samples were taken from different fractions of the extrudate. A fraction was defined as 1/6 of the total ink volume of the cartridge. The fractions were then dried in a vacuum oven for 2 h at 60 °C to remove residual free water and subsequently placed in aluminacrucibles. They were measured in the TGA apparatus by heating the sample from 50 °C to 900 °C with a temperature ramp of 5 °C/min. The weight was recorded as percentage of the initial sample weight at 50 °C.
2.2.9. Printing characterization
All printing was performed on a BioFactory bioprinter (regenHU, Switzerland). Designs for the printing were created using the BioCAD software (regenHU, Switzerland) or with Slic3r (http://slic3r.org/) in combination with a custom written MATLAB postprocessor. All samples were printed at room temperature and crosslinking was performed using a built-in UV-Pen (365 nm, 6.09 mW/cm2).
To assess the influence of printing speed on the strand size of ink from different compartments of the cartridge, single lines were printed at printing speeds between 50 and 1600 mm/min and their widths as well as heights were evaluated using ImageJ. This was performed for inks with 25% and 30% HAp content extruded with 0.61 mm conical and a 0.41 mm straight steel needle (Nordsen, USA). The extrusion pressures were chosen so that a continuous flow of ink from the cartridge was obtained. To show the suitability of the developed ink for biphasic constructs (e.g. osteochondral), ink with 0% HAp content was used for the top phase and ink with 25% HAp content was used for the bottom phase of the biphasic construct and both inks were printed on top of each other using a 0.41 mm straight steel needle. Inks contained 0.05% w/v Irgacure 2959. Passage 4 bovine chondrocytes were stained with CellTracker Blue (4-chloromethyl umbelliferone, 10 μM) for 30 min, washed with serum-free media and gently mixed into the 0% HAp ink at 10·106 cells/ml using a spatula. Printing of the HAp containing ink layer was performed at a pressure of 103 kPa and a speed of 215 mm/min. A total of 4 layers was printed. 2 layers with 0% HAp ink were then printed on top with a pressure of 25 kPa and a speed of 400 mm/min. UV-crosslinking was performed for 2 min after every printed layer and for an additional 5 min after the end of the top layer.
2.2.10. Interfacial bond strength
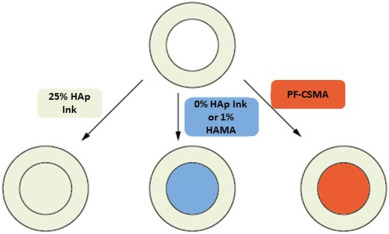
To assess the interfacial bond strength between two printed layers, an outer ring of 25% HAp content ink (representing the first printed layer) with an inner diameter of 4 mm was printed using a 0.61 mm conical needle, a pressure of 60 kPa and a printing speed of 400 mm/min. This first layer was then crosslinked for either 2, 4 or 8 min which simulates the curing process before deposition of the next layer. The ring was then filled manually (representing the second printed layer) with either 25% HAp ink, 0% HAp ink, 1% HAMA or 30% Pluronic-dimethacrylate mixed with 5% chondroitin sulfate-methacrylate (PF-CSMA) to assess the interfacial bond strength between different inks of a potential biphasic construct. To clarify the different combinations, we illustrated them in Fig. 1.
 Fig. 1. Different outer-inner layer configurations for the push-out tests.
Fig. 1. Different outer-inner layer configurations for the push-out tests.The interface between the two layers was then illuminated with UV-light with the UV source moving along the interface at a speed of 7.85 mm/min for a total of 2 min. After another 5 min of UV-exposure in the center of the second layer (representing the final crosslinking step after printing), the samples were placed in a sample holder and the push-out test was performed at 0.5 mm/s on a Texture Analyzer XT Plus (Stable Micro Sytems, United Kingdom). The sample holder consisted of a steel ring with an inner diameter of 5 mm and the probe head had a diameter of 3 mm. The force curve was recorded and the maximum force was divided by the interfacial area between the two layers to obtain the interfacial bond strength. Each interface area was calculated separately by quantifying the inner radius of the inner circle material with light microscope and carefully measuring the height of the printed sample using a calliper.
2.2.11. Cell adhesion and viability
The MSCs used in the adhesion and viability study were obtained from trabecular bone samples that were retrieved during surgical hip replacement of an otherwise healthy patient (male, 42 years) after having received informed consent. Approval of the protocol was obtained from the ethical board of the Kantonsspital St. Gallen, Switzerland (ethical committee approval number EKSG08/014/1B). The samples were incubated overnight at 4 °C in isolation medium (25 mM HEPES, 128.5 mM NaCl, 5.4 mM KCl, 5.5 mM D(+)-glucose, 51.8 mM D(+)-saccharose and 0.1% BSA). To remove fat tissue, the sample was centrifuged at 110 g for 15 min at 4 °C and the remaining pieces of trabecular bone were rinsed with isolation media several times. The solution was collected and filtered through a 200 μm cell strainer. The solution was centrifuged and the obtained cell pellet was resuspended in proliferation media (DMEM 31885, 10% FBS, 1% Pen-Strep and 5 ng/ml FGF-2). Passage 3 MSCs were used in the adhesion study.
The samples for the adhesion study were prepared in a rheometer setup by UV-crosslinking resulting in discs with a diameter of 20 mm and a thickness of 0.5 mm. Discs with a diameter of 6 mm were punched out using a biopsy punch and placed in sterile 100 mM CaCl2 overnight. 70% ethanol was then put in the wells for 10 min to disinfect the surface. This was followed by two washing steps with proliferation media. The discs were left to incubate in proliferation media for 1 h. The MSCs were then seeded on the ink disc containing different amounts of HAp at 40′000 cells/cm2 and cell adhesion was assessed after 3 days using a phalloidin-rhodamine/Hoechst staining. For the staining, the media was removed and the samples washed once with PBS and subsequently fixed with 4% formaldehyde in PBS for 25 min. After two more washes with PBS, the cells were permeabilized for 15 min using 0.1% Triton-X100 + 1% BSA. The solution was removed from the samples and they were rinsed 3 times with PBS. Phalloidin-rhodamine at a final concentration of 0.13 μM in serum-free media was then added to the samples for 30 min, after which Hoechst (10 μg/ml final concentration) was added and incubated with the samples for another 30 min. The samples were washed 3 times for 1 min with PBS and imaged with a fluorescent microscope (Zeiss Axio Observer, Zeiss, Switzerland).
For the viability measurements using live/dead staining, the seeded cells were washed once with media without FBS and subsequently stained with 1 μg/ml Calcein AM (1 μM) and 0.66 μg/ml Propidium Iodide (1 μM) for 15 min. This was followed by a single wash with cell culture media and the cells were then imaged with a fluorescent microscope.
To analyse the cell viability using the MTS assay, MTS stock solution was prepared according to the manufacturer's instructions (CellTiter 96® AQueous One Solution, Promega). MTS stock solution was then diluted to 0.04% in cell culture medium (150 μl medium with 30 μl MTS stock solution). Samples were incubated for 1 h and afterwards 150 μl of solution was placed in 48 well-plate and read out on a microplate reader (Synergy H1, BioTek) at 490 nm. Cell culture media was utilized as blanks and cells seeded on tissue culture plastic (TCPS) were used as positive and cells treated with 70% ethanol for 30 min were used as negative controls.
2.2.12. Scanning electron microscopy
For scanning electron microscopy (SEM), discs were created as described earlier using different amounts of HAp. To investigate if there is hydroxyapatite crystal formation on the gellan gum containing ink as previously reported elsewhere [34], the discs were placed in CaCl2 overnight before being exposed to cell culture media (without FBS) for 7 days at 37 °C in an incubator (5% CO2). Afterwards they were frozen at −80 °C and placed into a lyophilizer for freeze-drying. The freeze-dried discs were then coated with a 5 nm layer of platinum and subsequently analyzed in a Zeiss Gemini 1530 FEG SEM at 5 kV, at room temperature.
3. Results
3.1. Buffer mediated GG-MA synthesis
Robust synthesis methods that involve a minimal number of manual steps are essential for the reproducible production of large polymer batches to develop inks for 3D printing applications. As 3D printing often requires large volumes of ink (several ml) for large scaffold production, up-scaling of the synthesis is required. By utilizing 2-amino-2-methyl-1,3-propanediol (AMPD) as a buffer, we were able to eliminate the need to manually balance the pH at 8.5. Over the course of 3 h, the pH of the reaction only decreased from 8.5 to 8.33 and after 24 h we found a final pH of 7.91. Furthermore, the buffer mediated synthesis achieved a 2.5 times higher degree of substitution than the conventional synthesis method (Fig. 2a, red square) when a 20-fold molar excess of GMA was used in the reaction. The methacrylation of GG using GMA followed a linear trend up to 15-fold molar excess and batch-to-batch differences were small (1.76 ± 0.14% for the 5-fold, 1.72 ± 0.20% for the 5.6-fold and 3.14 ± 0.12% for the 10-fold excess, respectively). An increase to a 20-fold excess does not increase the DS, indicating that there is a kinetic limit of how many functional groups on GG can be modified within 24 h, independent on the excess of GMA.
 Fig. 2. a) Obtained degrees of substitution (DS) in buffer mediated GG-MA synthesis with varying fold-excess of GMA. A 2.5 higher DS was achieved with buffer mediated synthesis (black squares) compared to conventional synthesis (red square) at 20-fold molar excess. b) Influence of the synthesis method on the flow behaviour of inks with 25% HAp. The buffer mediated synthesis was performed with 5-fold excess GMA. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2. a) Obtained degrees of substitution (DS) in buffer mediated GG-MA synthesis with varying fold-excess of GMA. A 2.5 higher DS was achieved with buffer mediated synthesis (black squares) compared to conventional synthesis (red square) at 20-fold molar excess. b) Influence of the synthesis method on the flow behaviour of inks with 25% HAp. The buffer mediated synthesis was performed with 5-fold excess GMA. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)To evaluate the influence of the synthesis method on the properties of the ink, GG-MA produced by the conventional synthesis method (20-fold GMA excess) was compared with buffer synthesized GG-MA (5.6-fold GMA excess) in all experiments as those had the same degree of substitution. Rheological measurements were performed to assess the flow behaviour of inks produced with either synthesis method. As seen in Fig. 2b, the viscosity of an ink with 25% HAp content was decreased at all shear rates when the buffer mediated synthesized GG-MA was used. The same trend was found for ink containing 30% HAp (data not shown), indicating that the synthesis method for GG-MA indeed has an influence on the rheological behaviour of the produced HAp containing inks.
3.2. Influence of polymer processing
High molecular weight biopolymers are usually obtained from lyophilization as low-density, sponge-like materials. This complicates the creation of solutions with a high polymer content as complete hydration of these sponges with small amounts of water often proves difficult in our experience. We therefore decided to mechanically grind the obtained GG-MA and HAMA sponges into a powder using a cryomill to facilitate ink production. When inks with 30% HAp content and cryomilled polymers were rheologically assessed, we found differences in the viscosity of up to one order of magnitude (11.3 kPa·s at versus 113 kPa·s at a shear rate or 0.1 s−1) compared to the untreated polymers as shown in Fig. 3.
 Fig. 3. Influence of the cryomilling process on the rheological behaviour of an ink containing 30% HAp. In case of the untreated polymers, mixing was performed at 50 °C in a sealed vessel under constant stirring. The GG-MA in the ink was synthesized using the conventional synthesis method.
Fig. 3. Influence of the cryomilling process on the rheological behaviour of an ink containing 30% HAp. In case of the untreated polymers, mixing was performed at 50 °C in a sealed vessel under constant stirring. The GG-MA in the ink was synthesized using the conventional synthesis method.To evaluate if this difference is caused by a reduction in the biopolymers' molecular weights, SEC measurements were performed on the underivatized HA and GG, their methacrylated versions HAMA and GG-MA and the cryomilled HAMA and GG-MA. As shown in Table S1, Mw of HA increased with the addition of the methacrylate groups to the polymer from 516 to 534 kDa. The mass increase of 18 kDa correlates well with the added methacrylate groups at the obtained degrees of substitution. Upon cryomilling, the HAMA chains become fragmented which leads to a reduction of the Mw to 118 kDa and an increase in the polydispersity index (PDI) from 2.63 to 4.01. In case of GG, the addition of methacrylates reduced the Mw from 4587 to 4394 kDa which is much higher than expected from previous reports [35]. Cryomilling almost halved the Mw of GG-MA to 2542 kDa, indicating the mechanical disruption of the chains. Both the scission of the HAMA and GG-MA chains during cryomilling are in agreement with a reduction of the viscosities of the inks (Fig. 3).
3.3. Influence of HAp content on flow behaviour and UV-crosslinking
Particles in suspensions are known to introduce the possibility of shear thickening due to shear induced particle aggregate formation [36]. This effect would lead to undesirable clogging of the needle in a 3D printing application. To investigate the potential of the inks to undergo shear thickening, we assessed the flow behaviour of inks with 25% and 30% HAp content (Fig. 4a) using rotational rheology. The two concentrations were chosen based on maximizing the amount of HAp for high printing fidelity while still being able to print the material without clogging the nozzle. For comparison, the flow curve of a 0% HAp content ink is depicted. Shear thickening was not observed at any of the shear rates investigated for the particle containing inks. Fitting of the flow curve data using the Herschel-Bulkley equation [37] revealed a yield point of 357 ± 23 Pa and 516 ± 55 Pa for the ink with 25% and 30% HAp content, respectively. When the inks with 25% and 30% HAp content were mixed with the photoinitiator Irgacure 2959 and exposed to UV radiation (320–500 nm bandpass filter), crosslinking occurred and after 5 min the inks with 25% and 30% HAp content reached storage moduli of 50.5 and 82.4 kPa (Fig. 4b), respectively. Using equation Eq. (1) we can calculate the shear modulus.(1)
 Fig. 4. Flow behaviour of HAp containing inks compared to a 0% HAp ink (a) and changes in the storage modulus G' upon UV exposure (b) of inks with 25% and 30% HAp content. (c) Behaviour of the storage modulus G' of an ink with 25% HAp content upon repeated exposure to high shear.
Fig. 4. Flow behaviour of HAp containing inks compared to a 0% HAp ink (a) and changes in the storage modulus G' upon UV exposure (b) of inks with 25% and 30% HAp content. (c) Behaviour of the storage modulus G' of an ink with 25% HAp content upon repeated exposure to high shear.For the inks with 25% and 30% HAp content the calculated shear moduli were 50.6 and 82.9 kPa, respectively.
For an ink for extrusion printing, quick recovery after a high shear event such as extrusion through a needle is crucial. If the shear recovery of an ink is slow, it will continue to flow after deposition onto a substrate diminishing the resolution that can be achieved. Shear recovery experiments were therefore performed on the 25% ink to see if structural changes occur during shear and if the ink recovers quickly after the shear event. After the first high shear event we found that G' recovered to 45% of its initial value within 30 s as shown in Fig. 4c. After the second shear event, G' recovered to only 30%. Within 30 s after the third shear event, 27% of the initial G' value was obtained. These results indicate that repeated shear events induce structural changes in the ink.
3.4. Extrusion force measurements
While rheological analysis of inks for 3D printing can deliver information about their flow behaviour, it is not able to predict the extrusion performance through differently sized needle geometries. We therefore developed a setup which can be loaded with the same cartridges that are used in the printing process (Fig. 5a).
 Fig. 5. a) The extrusion force measurement setup consists of a plunger connected to the texture analyzer, the cartridge holder and the printer cartridge with the piston and ink. Inks with different HAp contents were extruded through. b) 0.61 mm conical needle, c) 0.41 mm conical needle and d) 0.41 mm straight steel needle. The 30% HAp sample is not shown as it was impossible to be extruded though a 0.41 mm straight steel needle. The insert in the subfigures b, c and d shows the needle type utilized for the extrusion force experiment. Dashed boxes in subfigures c and d highlight broad peaks from an unknown origin.
Fig. 5. a) The extrusion force measurement setup consists of a plunger connected to the texture analyzer, the cartridge holder and the printer cartridge with the piston and ink. Inks with different HAp contents were extruded through. b) 0.61 mm conical needle, c) 0.41 mm conical needle and d) 0.41 mm straight steel needle. The 30% HAp sample is not shown as it was impossible to be extruded though a 0.41 mm straight steel needle. The insert in the subfigures b, c and d shows the needle type utilized for the extrusion force experiment. Dashed boxes in subfigures c and d highlight broad peaks from an unknown origin.With this setup we were able to measure the extrusion force required for different needle geometries and ink compositions. Furthermore, with this method it is possible to assess the quality of the ink mixing process and cartridge filling. Inhomogeneous inks are revealed by a constant fluctuation of the extrusion force with aggregates being indicated by a positive peak whereas, on the other hand, air introduced during the cartridge filling procedure is indicated by a negative peak in the extrusion force. When inks with 0, 25 and 30% HAp content were extruded from the printer cartridge equipped with a 0.61 mm conical printing needle (Fig. 5b), the extrusion force was the highest for the ink with 30% HAp content. The extrusion force decreased with decreasing amounts of HAp particles in the ink. This was expected as the viscosities of the inks also decrease with decreasing HAp content (Fig. 4a). The ink with 30% HAp content displayed positive peaks in the recorded force curve indicating the presence of larger aggregates in the ink that cause short flow instabilities which hinder continuous printing. In the ink with 25% HAp content, no positive peaks were observed. However, we observed a negative peak which represents an air bubble trapped during cartridge filling. The same ink without any HAp (0% HAp content) did not display any peaks. Compared to the 0.61 mm conical needle, extrusion forces for all inks increased when extruded from a cartridge equipped with a smaller conical needle of 0.41 mm. In the case of the ink with 30% HAp content, positive peaks frequently occurred as shown in Fig. 5c. This means that aggregates with sizes in range of the inner needle diameter are present in the ink. The 30% HAp ink is therefore unsuitable for printing with a 0.41 mm conical needle. In the case of the ink with 25% HAp content, no bubbles or peaks were observed apart from a very broad peak between 38 and 53 s (dashed box in Fig. 5c). A broad peak of similar shape was observed for the 25% HAp ink extruded through a straight 0.41 mm steel needle (dashed box in Fig. 5d) and it is currently unclear if it is caused by aggregates or an artefact from the measurement setup. In the case of the 0% HAp ink, extrusion through a 0.41 mm conical needle was possible without any clogging and no bubbles were introduced during the cartridge filling process. The sharp increase in the extrusion force indicates that all the ink contained in the cartridge was extruded and the plunger hit the bottom of the cartridge. This happened earlier for the 0% HAp inks as smaller volumes of 0% HAp ink was utilized compared to the 25% and 30% HAp inks. To see if there are differences in the extrusion behaviour of inks between conically shaped and straight needles, they were extruded through a 0.41 mm straight steel needle (Fig. 5d). Due to the fact that the 30% HAp ink was already considered unsuitable to be printed using a conical 0.41 mm needle, it was excluded from the experiment. Compared to the conical needle, both the inks with 0% and the 25% HAp content required a higher force to be extruded through the straight needle. The 0% HAp content ink did not show any clogging behaviour and the same was true for the 25%. However, there was a broad peak between 24 and 28 s with a similar shape as the broad peak in Fig. 5c but with a smaller peak size compared to the overall extrusion force. We also tested if the clogging observed during the extrusion is based on pre-existing agglomerates or originates from agglomerates which formed during the ink preparation process. To do so, we sieved the HAp particles through a sieve with a 40 μm mesh. As is shown in Fig. S3a, sieving of the particles before the ink preparation lowered the extrusion force for the inks but did not prevent the aggregates altogether, indicated by the frequent appearance of positive peaks during the measurement. However, sieving of the particles enabled to create inks with 30% HAp content which did not clog the needle completely when extruded, indicating the removal of larger aggregates (Fig. S3b). From the obtained data we concluded that the 0% and the 25% HAp content inks are suitable for printing with both the conical and straight 0.41 mm needles, whereas the 30% HAp content ink would only be suitable to be printed with the conical 0.61 mm needle.
3.5. Ink homogeneity
To confirm a homogeneous distribution of HAp particles, we additionally performed TGA measurements to evaluate the solid particle content in different parts of the cartridge (Fig. 6a). As shown in Fig. 6b, TGA measurements revealed that, despite drying of the samples in a vacuum oven at 60 °C, there was still residual water (most probably bound) in the sample which evaporated in the temperature window between 50 °C–200 °C (2.5% of the initial weight). Between 200 °C–900 °C, a loss of 10.9–11.3% of the initial weight was observed, which is equivalent to the total dry polymer mass included in the ink. The residual 86.2–86.6% at 900 °C represent the HAp particles. As evident from the TGA curves and from the insert, there was only a marginal difference between the sections within the printer cartridge in terms of the composition.
 Fig. 6. a) Schematic of the compartments within the printer cartridge which were evaluated with TGA. b) TGA measurement of different sections of a cartridge loaded with 30% HAp content ink. The insert shows a magnified view of the weight differences between 850 °C and 900 °C.
Fig. 6. a) Schematic of the compartments within the printer cartridge which were evaluated with TGA. b) TGA measurement of different sections of a cartridge loaded with 30% HAp content ink. The insert shows a magnified view of the weight differences between 850 °C and 900 °C.3.6. Ink printing
To evaluate the influence of printing speed on the strand size, lines were printed from inks with different HAp content using a range of printing speeds. The ink with 30% HAp content was printed using a 0.61 mm conical needle (Fig. 7a) and the ink with 25% HAp content was printed with a 0.61 mm conical (Fig. 7b) and a 0.41 mm straight steel needle (Fig. 7c) as these were found to be suitable for printing without any clogging. The lines were evaluated at three different locations and an average was taken from two lines. For the 30% HAp content ink, we found differences in the thickness of the lines when printed with ink originating from different compartments of the cartridge (Fig. 6a). However, the effect was only present at lower printing speeds (800 mm/min, Fig. 7a) and diminished at higher speeds. The effect was less pronounced for the 25% HAp ink (Fig. 7b) when printed with a 0.61 mm conical needle. Differences were especially minimal at higher printing speeds (>600 mm/min).
 Fig. 7. Line thickness at different printing speeds. a) 30% HAp content ink, 0.61 mm conical needle, printed at 95 kPa b) 25% HAp content ink, 0.61 mm conical needle printed at 55 kPa pressure and c) 25% HAp content ink, 0.41 mm straight steel needle printed at 105 kPa pressure. d) Width/height ratio average over the different printing speeds. The error bar indicates the variation of the width/height ratio between when using different printing speeds with the same needle.
Fig. 7. Line thickness at different printing speeds. a) 30% HAp content ink, 0.61 mm conical needle, printed at 95 kPa b) 25% HAp content ink, 0.61 mm conical needle printed at 55 kPa pressure and c) 25% HAp content ink, 0.41 mm straight steel needle printed at 105 kPa pressure. d) Width/height ratio average over the different printing speeds. The error bar indicates the variation of the width/height ratio between when using different printing speeds with the same needle.When a 0.41 mm straight steel needle was used for the extrusion of the 25% HAp ink, the printing pressure had to be increased to obtain a continuous flow of ink. The reason for this is the sharp transition from a large to a small diameter where the ink has to pass through. To minimize the effects of die swell (which increases with increasing pressure and flow rate of the ink), the lowest possible pressure for extrusion was chosen and printing was performed at low speeds (50–150 mm/min). There were still differences between the different parts of the cartridge (Fig. 7c) but they did not follow a clear trend. Because the line thickness was less influenced by the ink's origin from within the cartridge, we chose to use the 25% HAp ink for all future experiments involving printing. We also compared the ratio between the height and the width of the produced strands at different printing speeds (Fig. 7d). The lower height/width ratio of the 25% HAp ink indicates that it continues to flow for a longer period of time following deposition, which means it recovers slower after shear, compared to the 30% HAp ink. When comparing the height/width ratio of the 25% HAp ink printed with either the 0.61 mm conical or the 0.41 mm straight steel needle, less variation was found between different printing speeds in the case of the 0.41 mm straight steel needle. Relaxation experiments on all the different inks revealed that with increasing HAp content, the stress within the materials (evident as normal force) is increased and that relaxation processes are slower (Fig. S4). As the lines printed with the 0.41 mm straight steel needle were more homogeneous and less dependent on printing speed, we chose this setup for all future printing experiments to ensure consistent printing.
To assess the printability of the 25% HAp ink with the ideal needle geometry for this ink, cylinders with varying infill patterns were generated in Slic3r and postprocessed for the Biofactory in a custom written matlab postprocessor (Fig. S5). Printed structures corresponded well with the STL design, having fully interconnected pores. Sharp corners however became more rounded caused by the speed at which the printer was moving.
To demonstrate the possibility of printing a bisphasic construct in a single process, grids of 10 × 10 mm were printed using the ink with 25% HAp content and the ink with 0% HAp containing cells (Fig. S6). The chosen interline spacing was 1.5 mm which resulted in pores with an average diameter of 794 ± 65 μm, which is larger than the minimal pore size utilized for example in bone engineering [38]. For the bottom layer, 4 layers were printed up to a total height of 1.86 mm and horizontal pores were visible, which indicates that free standing strands could be produced with the 25% HAp content ink.
Because we used a 3D printing approach to create biphasic constructs, it was possible to print the cell containing layer directly on top of the bottom layer containing 25% HAp. This circumvents the need to post-assemble the two layers in a separate step. The top layer consisting of the ink with 0% HAp content and containing the bovine chondrocytes nicely attached on top of the bottom layer after UV-crosslinking (Fig. S6). The labelled bovine chondrocytes were visible by fluorescence microscopy and a clear interface between the two layers was observed (indicated by a dashed white line in Fig. S6).
3.7. Interlayer adhesion forces
A push out test was performed to assess the bond strength of the interface between two printed ink layers containing 25% HAp (Fig. 8a) as well as the bond strength between the inks containing 25% and 0% HAp (Fig. 8b). For the push-out test, an outer ring of ink with 25% HAp content was printed and crosslinked for either 2, 4 or 8 min. The ring was then filled manually either with 25% HAp ink, 0% HAp ink, 1% HAMA or 30% Pluronic-dimethacrylate mixed with 5% chondroitin sulfate-methacrylate (PF-CSMA) followed by subsequent crosslinking via exposure to UV-light. The filling was then pushed out and the force required to break the interface was recorded. The push-out test revealed that there was no significant difference in the interfacial bond strength between outer ring and the filling if the outer ring was UV-crosslinked for 2 or 4 min (27.5 ± 5.3 kPa and 24.7 ± 2.9 kPa, respectively) before the filling was added and UV-crosslinked for another 2 min at the interface. After crosslinking the outer ring for 8 min, however, there was a significant drop (Oneway ANOVA, Bonferroni post-hoc, p < .05, n = 4) in the interfacial bond strength (18.5 ± 3.0 kPa), indicating that the amount of available methacrylate groups on the surface of the outer ring was diminished. The amount is reduced to an extent where fewer methacrylate groups are available than could theoretically be crosslinked in the 2 min of UV-crosslinking at the interface.