1. Introduction
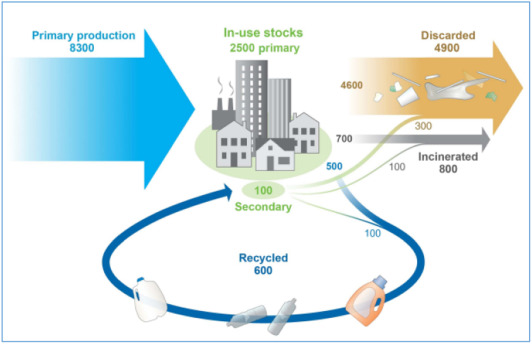
Over the past few decades, negative environmental impacts caused by petroleum-based polymers coupled by the uncertainty of crude oil prices and depleting petroleum resources has been a major concern. At the end of 2015, it was estimated that 8300 million metric tons (Mt) of virgin petroleum based polymers for resins, fibers, and additives were produced in eight industrial sectors. Nearly 6300 Mt of plastic waste had been produced, of which 12% was incinerated, 9% was recycled, and 79% was gathered in landfills or the natural environment, as shown in Fig. 1 [1]. In addition, the main global source in 2015 was PP and PE, contributing 46% to the global production and 63.4% in packaging industry as illustrated in Fig. 2. Based on the present production and waste management trends, approximately 12,000 Mt of non-biodegradable polymers waste were expected to be disposed of in the landfill by 2050 [2].

Fig. 1. Global production, use, and fate of polymer resins, synthetic fibers and additives from 1950 to 2015; in million metric tons.
Copyright (2017) Science Advances [1].
Fig. 2. (a) Types of plastics produced worldwide (in percent), and (b) Plastic use in the packaging industry in the world in 2015.
Copyright (2017) Royal Society of Chemistry [2].Realizing the downside of these unfavorable polymers, many countries have initiated strategies and policies targeting this global issue, for instance, Germany and Denmark were among the first to ban single-use plastic bag in most retail stores back in the 1990s. Subsequently, countries in Africa, Asia, Ireland, and the rest of Europe gradually introduced bans or enforcement of plastic bag consumption tax and levy; which contributed to reducing the overall consumption significantly [3]. To address the root cause of environmental pollution, a viable approach is to use bio-related polymers as substitutes to replace petroleum-based polymer, largely due to their excellent eco-friendly attributes.
One of the most promising biopolymers is polyhydroxybutyrate (PHB), a biogenic short chain polyhydroxyalkanoate (PHA) polymer produced by fermentation process; first discovered by Lemoigne in the 1920s as an intracellular energy and carbon storage material accumulated in various microorganisms, such as bacteria Alcaligenis euterophus, Bacillus, and Pseudomonas [4], [5]. PHB exhibits remarkable mechanical properties, comparable only to 100% meso diads-polypropylene (PP) and polyethylene (PE). Fig. 3 shows the chemical structure of PHB. Its exceptional stereo-chemical regularity of the structure leads to a highly crystallized homopolymer with crystallinity up to 70%, contributing to its excellent mechanical properties; high elasticity modulus of around 2.5–3 GPa and tensile strength at break of 35–40 MPa. In addition, the lamellar structure contributes to superior gas barrier properties with its water vapor permeability at about 560 g μm/m2/day, making it suitable for low-end food packaging applications [6], [7], [8], [9], [10], [11], [12], [13], [14], [15].
 Fig. 3. Chemical structure of PHB.
Fig. 3. Chemical structure of PHB.Furthermore, PHB is biodegradable and biocompatible, which in turn dictates its environmental fate in terms of ecotoxicity and human toxicity [16]. As such, PHB has found valuable applications in tissue engineering and other biomedical related applications, such as surgical sutures, thermogels as a controlled-release drug delivery vehicle, surgical meshes, wound dressing and absorbable nerve guides, tissue scaffolding for bone and nerve regeneration, cardiovascular and cartilage support [17], [18], [19], [20], [21], [22].
While PHB has many desirable properties, its widespread applications have been limited due to myriad challenges, which includes (1) inherent physical aging effect, based on secondary crystallization which lead to embrittlement[23], (2) slow crystallization rate and low nucleation density promotes formation of large spherulites lead to easier crack and fracture, evident from the low elongation at break of 5–7% [24], (3) thermal instability due to narrow thermal processing window, where PHB degrades via random chain scission on the ester bond in the temperature range of 170–200 °C [25], [26] and lastly, (4) high production cost which limit its competitiveness in industrial and commercial applications [27], [28], [29].
Many strategies have been devised to toughen PHB such as modification through drawing and thermal treatment, blending with materials from natural sources and synthetic polymers with suitable molecular structures; inclusion of natural fibers or rigid fillers to form reinforced composites and lastly, modification by chemical functionalization as illustrated in Fig. 4. The enhancement in toughness and flexibility are normally at the expense of material stiffness and strength [30], [31], [32], [33], [34]. Therefore, mutual enhancement of these properties has been a grand challenge thus far and currently, this challenge has attracted the interest of many research groups. However, there are limited reviews on PHB toughening. In this contribution, we intend to provide a comprehensive review of PHB toughening materials including the future perspectives of PHB biopolymer. Table 1 is a summary of various toughened PHB systems which show promising applications in construction, packaging and biomedical applications.
 Fig. 4. Graphical illustration of the different PHB modification approaches.
Fig. 4. Graphical illustration of the different PHB modification approaches.Table 1. Material characteristic properties of toughened PHB via different strategies and their applications.
| Strategies | Material type | Enhanced thermal and mechanical features | Application(s) | Ref |
|---|---|---|---|---|
| Drawing and thermal treatment | PHB film |
1st drawing (DR: 10): σ = 100 MPa, εb = 67%, 2nd drawing (DR: 1.5): σ = 388 MPa, E = 2.94 MPa |
Marine, agricultural and medical | 38 |
| UHMW-PHB fiber | 2-step drawing (DR: 60): E = 8.1 GPa, σ = 1.3 GPa, εb = 35% | Fishing line and suture | 39 | |
| UHMW-PHB/PHB fiber | 1-step drawing (DR: 12): E = 1.5–2.51 GPa, σ = 211–242 MPa, ɛb = 74–88%, Tm = 170 °C | – | 40 | |
| PHB/ENR film | Tg = − 14.1 °C | – | 42 | |
| PHB/P(E-MA-GMA) film | ISunotched = ~ 1100 J/m, εb = 14–17%, σUTS = 17 MPa, E = 1.2–1.3 GPa | Agricultural and food packaging | 43 | |
|
PHB/P(E-MA-GMA)/TEC film PHB/P(E-MA-GMA)/TBC film |
ISunotched = ~ 180 J/m, εb = 2.2%, σUTS = 9.4–11 MPa, E = 0.8–1.1 GPa, Tg ~ 19 °C | Agricultural and food packaging | 44 | |
| PHB/TBC/BN fiber | σUTS = 215 MPa, εmax = 40% | Textile manufacturing and medical | 45 | |
| Blending | PHB/soluble potato Starch | Tg ~ 63–87 °C, Tm = 165–168 °C, σUTS = 31.5 MPa | Paper coating & cardboard packaging | 46 |
| PHB/maize starch | Tg ~ 37 °C, Tm = 165–168 °C, σ = 31.5 MPa | – | 47 | |
| PHB/destructured starch | σ = 20 MPa, εb = 0.8%, E = 4 GPa | Medical | 488 | |
| PHB/maize starch | σ = 18.7–22.0 MPa, εb = 2.62%, E = 0.37–1.32 GPa | Agricultural & marine | 49 | |
| PHB/starch |
Tg,1 = − 58.1 °C, Tg,2 = − 19.2 °C, Tm = 141–159.1 °C, σ = 3–11.3 MPa, ε = 2.3–15.5%, E = 0.12–0.21 GPa |
– | 50 | |
| PHB/starch/glycerol | σ = 5.9 MPa, Tear strength = 45 kJ/m2 | – | 51 | |
| PHB/EVA/starch/MA | σ = 15–20 MPa, εb = 50%, Work = 0.09 Nm | – | 52 | |
| PHB or PHB-g-AA/starch | Tm = 167.8–171.3 °C, σ = 17.5 MPa | Agricultural, marine & medical | 53 | |
| PHB/PVAc modified starch | Tm = 160–170 °C, σ = 6.4–15.4 MPa, εb = 2.9–21%, E = 0.33–1.06 GPa | – | 54 | |
| PHB/cellulose nanocrystals (CNC) | Tm = 150–172.5 °C, σ = 20–30 MPa, εb = 1.4–2.2%, E = 3.5–4 GPa | Packaging | 55 | |
| PHB/cellulose nanowhiskers (CNW) | Tm = 156–163 °C, σ = 17 MPa, εb = 300%, E = 0.1–0.6 GPa | Medical & agricultural | 56 | |
| PHB/cellulose/DCP | Tm = 155–171 °C, σ = 25.9 MPa, εb = 13.2%, E = 5.5 GPa | Packaging and semistructural profiles | 57 | |
| PHB/bacterial cellulose (BC) | εmax = 10.5%, σmax = 91.4 MPa | Tissue engineering and medical | 58 | |
| PHB/hydroxethyl cellulose acetate | Tm = 155–175 °C | – | 60 | |
| PHB/ethyl cellulose (EC) | Tm = 170–175.5 °C | – | 61 | |
| PHB/ethyl cellulose (EC) | IS = 7 kJ/m2, εb = 1.5%, σ = 33.3 MPa, Tm = 163.6–175.9 °C | Biotechnology | 62 | |
| PHB/CAP and PHB/CAB | εb = 0–5%, σ = 2–10 MPa | – | 63 | |
| PHB/insoluble lignin/or holocellulose | Tm = 172–175 °C | Diverse applications | 66 | |
| PHB/crude lignin and PHB/soda lignin | Tm = ~ 143.9–175 °C | Agriculture, cosmetics and packaging | 67 | |
| PHB/lignin | Tm ~ 151–174 °C | Packaging & pharmaceutical | 68 | |
| PHB/chitosan | εb = 43.3–82.9%, σ = 3.4–4 MPa, E = 4.9–8.7 MPa | Tissue engineering | 69 | |
| PHB/chitosan/or α-chitin | Tm ~ 155–170 °C | Biomedical | 70 | |
| PHB/chitin | Tm ~ 166–170 °C | Biomedical, agricultural, and paints | 71 | |
| PHB/PHBV film | E′ = 3.8–4.2GPa | – | 72 | |
| PHB/PHBHHx film | εb = 106–120%, Tc ~ 50 °C | Tissue engineering | 74 | |
| PHB/PLA/TBL | Tm ~ 165–167 °C, σ = 9–14.4 MPa, ε = 12.5–193.2%, E = 0.07–0.5 GPa | – | 84 | |
| PHB/PDLLA | Tm ~ 147–165 °C, σ = 35–45 MPa, σFlex = 45–65 MPa, EFlex = 2–3.5 GPa, IS = 15–35 kJ/m2 | Medical | 85 | |
| PLA/PHB | Tm ~ 155–165 °C, σ = 27.5–37.5 MPa, ε = 5–20% | Medical | 86 | |
| PHB/Sc-PLA | Tm ~ 210–230 °C | Medical devices | 87 | |
| PHB/PCL film | εb = 7.09%, σ = 1.43 MPa, E = 7.3 MPa | Drug delivery and biomedical | 96 | |
| PHB/PCL composite |
IS = 10.6 J/m2, εb = 11.2–1000%, σ = 17.3–21.4 MPa, E = 690–1643 MPa, EFlex = 716–1605 MPa, σFlex = 27–38.5 MPa, Tm = 171.2–172.7 °C |
Food packaging | 97 | |
| PHB/PCL/DCP composite | εb = 21.3%, IS = 6.3 kJ/m2, Tm = 55.5 °C | Food packaging | 98 | |
| PHB/PPC/PC film | IS = ~ 45 kJ/m2 | Biomedical | 101 | |
| PHB/PPC/PVAc/ATEC films | εb = 335–475%, Tc = 63 to 85 °C, Tg = − 8 °C | Air and water filter, wound dressing, drug delivery, surgical suture and mat fiber used in children diapers | 102 | |
| PHB/PBS/DCP film |
IS = 3.5–5.5 kJ/m2, ISunotched = 17–82 kJ/m2, ɛb = 4–15%, σflex,yield = 54–60 MPa, σys = 37–40 MPa |
Diverse applications | 103 | |
| PHB/PCL-b-PEG | εb = 350–382%, Tm,1 = 174.8–177.9 °C, Tm,2 = 157–159.4 °C | Biomedical | 107 | |
| PHB/PEO | Ti,1 = 216–239 °C, Ti,2 = 351–357 °C | Biomedical | 108 | |
| PHB/PVA film | εb = ~ 210%, Tm,1 = 178–186 °C, Tm,2 = 123–132 °C | Packaging, agricultural and biomedical | 110 | |
| PHA/PVAs/peroxide composite | εb = ~ 211–557%, IS = 21.3–42.7, tear strength = 10–80 g/mil, E = 494–1241 GPa, σb = 21.1–24.5 MPa | Automotive, consumer durable, construction, electrical, medical, packaging products | 113 | |
|
PHB/NR/MP composite PHB/EPR/MP composite |
IS = 124 J/m | Structural |
114, 115 |
|
| PHB/PP composite | IS = 22.5–29 J/m, εb: 4.5% E = 1.5–1.88 MPa, σ = 24.5–27.5 MPa, shore D hardness = 61–65, Tm = 177–180 °C | – | 118 | |
| PHB/PP-PHB/manganese stearate composite | εb = 8%, σ = 35 MPa. EFlex = 2.32 GPa, σFlex = 50 MPa | Cosmetic containers; cell phones; laptops; and packaging | 120 | |
|
PP/PHB/compatibilizer* *PP-MAH, P(E-MA), P(E-GMA) & P(E-MA-GMA) |
IS = 80 J/m, εb = ~ 80% | – | 121 | |
| LDPE/PHB film | Tm,1 = 105.9–106.8 °C, Tm,2 = 171.8–173.2 °C | Packaging materials | 123 | |
| PHB reinforced composite | PHB/flax fiber/TDP | Tm = 168–172 °C, E′ = ~ 13 GPa | Automotive (door and rear-parcel shelf panels) | 125 |
| PHB/flax | E = 8 GPa, σ = 40 MPa, IS = 135 J/m | Automotive | 126 | |
| PHB/agave | IS = 34.4 J/m, σ = 14 MPa, E = 770 MPa, EFlex = 2154 MPa, σflex = 17 MPa, E′ = 3733 MPa, Tg = 11.1C, Td = 25.2 °C | Construction | 128 | |
| PHB/Hemp fabric | σwarp = 55.9 MPa, εb,weft = 9%, Ewarp = 5.47 MPa, EFlex = 5.05 GPa, Tm = 117 °C | Construction | 129 | |
| PHB/jute, PHB/hemp, PHB/lyocell | σmax = 17 MPa, EFlex = 4 GPa, IS = 0.25 J | Biomedical | 130 | |
| PHB/carnauba fibers | Tm = 117 °C, σUTS = 27.5 MPa, EFlex = 3.25 GPa | Automotive | 131 | |
| PHB/cellulose fibers/DBEEA | Tm = 158.1 °C | Packaging | 132 | |
| PHB/curaua fibers/TEC | Tm = 117 °C, σUTS = 25 MPa, εmax = 14%, E = 178 MPa, IS = 70 J/m | Automotive and appliances | 133 | |
| PHB/SWCNTs | E = 11.74 GPa, hardness = 0.35 GPa | Tissue engineering | 136 | |
| PHB/acid-treated or alkylated MWCNTs | Tm = 171–177C, E′ = 4000–5200 MPa | Medical and pharmaceutical | 137 | |
| PHB/NaMMT or OMMT (30B) | E = 3.44 GPa, σ = 27 MPa | Biomedical, agricultural, and packaging | 138 | |
| PHB/C18MMT | E′ = 552 MPa, Tg = 29.5 °C, Tm,1 = 156.4 °C, Tm,2 = 166.13 °C | Tissue engineering | 139 | |
| PHB/OMMT | E = 15.5 MPa, σ = 10 MPa, εb = 0.72%, Tm,1 = 172.9 °C, Tm,2 = 169.3 °C | Packaging | 140 | |
|
PHB/A-fnSiO2 PHB/GNP |
Tm,1 = 158 °C, Tm,2 = 167 °C, pencil hardness = HB | Tissue engineering, medical devices and food packaging | 141 | |
| PHB/MGO | E = 83.6–114.6 MPa, σ = 20.7–37.4 MPa | Electrical | 142 | |
| Chemical modification | PHB-co-HV | εb = 970%, Tg = − 9 °C | – | 106 |
| PHB-co-HHx film | εb = 850%, Tg = − 4 °C | – | 148 | |
| PHB-co-HHx film |
1st drawing (DR = 50): σ = 80 MPa, εb = 258%, E = 0.87 GPa 2nd drawing (DR = 25): σ = 150 MPa, εb = 110%, E = 1.5 GPa |
– | 149 | |
|
PHB-g-ETA PHB-g-MAc |
Ti = 223.4 °C, Td = 270.3 °C, Tm = 165 °C | Biomedical | 150 | |
| PHB-co-PEG urethane | εb = 1912%, σys = 10.2 MPa, σb = 10.7 MPa, E = 120 MPa, Ti = 227 & 350 °C | Biomedical | 151 | |
| PHB-co-PEG urethane |
εb,dry state = 1090%, εb,hydrated state = 1962%, Edry state = 616.4 MPa, Ehydrated state = 342.7 MPa, σb,dry state = 18 MPa, σb,hydrated state = 14.5 MPa |
Tissue engineering | 152 | |
| PHB-co-PCL-PEG-PCL urethane | εb = 280–448%, σUTS = 9.2–10.5 MPa, Tm,1 = 126–131 °C, Tm,2 = 139–141 °C, Ti = 203–228 °C | Biomedical | 153 | |
| PHB-co-atactic PHB | Tm = 145.4 °C, Tg = − 2.2 °C | Cardiovascular engineering | 154 | |
| PHB-co-PLA-co-PCL | Tg,1 = − 31.2-23.5 °C, Tg,2 = 11.2–32.8 °C, Tm = 123.4–130.2 °C | Biomedical | 155 | |
| PLA-co-PHB-co-PLA | εb = 21%, Tg = 4.8 °C, Tm = 208.9 °C | Packaging and drug delivery | 156 |
PHB: polyhydroxybutyrate, UHMW-PHB: ultrahigh molecular weight polyhydroxybutyrate, ENR: epoxidized natural rubber, P(E-MA-GMA): poly(ethylene-co-methyl acrylate-co-glycidyl methacrylate), TEC: triethyl citrate, TBC: tributyl citrate, BN: boron nitrate, MA: maleic anhydride, AA: acrylic acid, DCP: dicumyl peroxide, PHBV or PHB-co-HV: poly(3-hydroxybutyrate-hydroxyvalerate), PHBHHx or PHB-co-HHx: poly(3-hydroxybutyrate-co-3-hydroxyhexanoate), TBL: tributyrin, PLA: polylactic acid, PDLLA: poly-dl-lactic acid, PCL: polycaprolactone, PPC: poly(propylene carbonate), PC: polycarbonate, PVAc/ATEC: polyvinyl acetate/acetyl triethyl citrate, PBS: polybutylene succinate, PEG: polyethylene glycol, PEO: polyethylene oxide, PVA: polyvinyl alcohol, EPR: ethylene-propylene rubber, NR: natural rubber, MR: maleated rubber, MP: maleated polybutadiene, PP: polypropylene, HDPE: high density polyethylene, PA6: polyamide 6, ABS: acrylonitrile butadiene styrene, PP–MAH: poly(propylene-g-maleic anhydride, P(E-MA): poly(ethylene-co-methyl acrylate), P(E-GMA): poly(ethylene-co-glycidyl methacrylate), DBEEA: bis[2-(2-butoxyethoxy)ethyl] adipate, SWCNTs: single-walled carbon nanotubes, MWCNT: multi-walled carbon nanotubes, NaMMT: natural montmorillonite, OMMT: organo-modified montmorillonite, A-fnSiO2: amino-functionalized nano-silica, GNP: graphene nano-platelets, MGO: modified graphene oxide, ETA: exo-3,6-epoxy-1,2,3,6-tetrahydrophthalic anhydride, MAc: maleic acid. DR: drawing ratio, ɛb: elongation at break, σ: tensile strength, σb: tensile strength at break, σUTS: ultimate tensile strength, σFlex: flexural strength, E: Young's modulus, EFlex: flexural modulus, E′: storage modulus, Tm: melting temperature, Tg: glass transition temperature, Ti: onset decomposition temperature, Td: decomposition temperature.
2. PHB toughening through drawing and thermal treatment
Crystallization and the size of the crystals have a great impact on the mechanical and thermal properties of polymers. PHB's exceptional stereochemical regularity and slow nucleation density results in the formation of very large spherulites which promote inter-spherulitic cracking. In addition, secondary crystallization ascribed to crystal perfection occur over time, where the amorphous segments are restricted in between the crystalline segments lead to a lamellar structural reorganization, causing embrittlement which severely reduces its mechanical performance [35]. Therefore, the combination of drawing which remodels the molecular chains orientation along the drawing direction and thermal annealing at elevated temperatures and re-aging at room temperature could expunge secondary crystallization, improving overall toughness and ductility [36], [37]. In this section, various formulation of PHB that were drawn and annealed are discussed.
For example, a group of researchers from RIKEN Institute reported a series of novel drawing and annealing processes to mitigate negative effects of aging and restore toughness by tuning the draw ratios and annealing temperatures. The resulted ultra-high-molecular-weight PHB film (UHMW-PHB) which was drawn by uniaxial one-step loose hanging weight method at around 160 °C silicone oil, followed by annealing at 160 °C for 2 h showed a remarkable increase of elongation at break from 6 to 67% and tensile strength from 77 to 100 MPa but at the expense of Young's modulus as it decreased from 2.3 to 1.8 GPa [38]. Amazingly, the properties were maintained even after 6 months with a higher Young's modulus at 2.5 GPa, tensile strength at 100 MPa and elongation at break remain unchanged. After which, the authors reported that film annealed at low temperature (100 °C for 2 h) displayed higher tensile strength while high temperature (160 °C for 2 h) displayed higher Young's modulus. Second drawing and annealing further increased the tensile strength and Young's modulus to about 388 MPa and 2.94 MPa respectively, accompanied by 25% decrease in elongation [38].
The same research group further improvised the process by using a lab-scale extruder to melt spin the fibers, followed by a combination of cold-drawing using iced water at 4 °C, second drawing at room temperature and a final thermal annealing process to increase the overall crystallinity. The two-step drawn (600% + 1000%) and annealed fiber showed an overall enhancement compared to as-spun native PHB, reaching a tensile strength of 1.3 GPa, elongation at break of 35% and Young's modulus of 18.1 GPa. The strengthening effect was due to the formation of β-form lamellar crystals being restrained in the 2D amorphous chains between α-form (21 helix conformation) crystals, generating a planar zigzag conformation which increased the polymer chains alignment, contributing to the high mechanical properties as shown in Fig. 5[38], [39].

Fig. 5. Schematic illustrations showing the proposed mechanism for generating β-form structures in blend and PHB films. (A) Initial crystallization of lamellar crystals (α-form). (B) Generating the β-form in an amorphous region during further crystallization. (B′) Movement of lamellar crystals during further crystallization. The β-form was formed from tie-chains fixed between lamellar crystals during annealing.
Copyright (2012) American Chemical Society Macromolecules [40].Kabe et al. fabricated annealed melt-spun fibers of PHB with ultra-high-molecular-weight PHB (UHMW-PHB) at the composition of 5/95 and 10/90 w/w% by similar cold-drawing methods. The annealed fiber of the 5/95 blend showed improved performance in tensile strength, Young's modulus, and elongation at break achieving 242 MPa, 1.50 GPa, and 88% respectively, comparable to poly(ethylene terephthalate) (PET). The wide-angle X-ray diffraction results indicated that UHMW-PHB act as nucleating agent and addition of small amount caused the formation of β-form in blend films. [40]The combined process of fiber drawing and annealing also showed a significant effect on the strengthening of PHB-co-HHx fiber [41]. In addition, Lee et al. showed that annealing allows two immiscible blends to bind together, obtaining a single Tg. In this work, the authors utilized PHB and epoxidized natural rubber (ENR-50), in which the reaction mechanism involved carboxyl end groups of degraded short PHB chains reacting with epoxide group of the ENR-50 [42]. Kurusu and his co-workers [43] investigated the effect of annealing on mechanical properties of pure PHB and its blends with poly(ethylene-co-methyl-acrylate-co-glycidyl-methacrylate) (PEMAGMA). Aging occurred in the non-annealed sample just after 18 h, showing a drastic difference in mechanical properties between the sample at 6 h and 1 day. Similar observations were revealed in the DSC and SAXS analysis, where the melting endotherms of the samples presented a single peak, 24 h after processing [43].
This clearly indicated that the second phase, PEMAGMA, acted as a nucleating agent. The same authors further investigated the effect of annealing on the injected molded samples under two conditions (110 °C for 10 min and 125 °C for 1000 min) and proposed a toughening mechanism [43]. It was revealed that sample aging resulted in thinner lamellae and imperfect crystals between stable crystals, with a more interfacial area between crystalline and amorphous regions as shown in Fig. 6. Annealing treatment reduced the chain density in the amorphous regions, the defective crystals in the melt, whereas, the stable crystals become thicker with increasing L, lc and la, enhancing the ability to dissipate energy [43]. Longer annealing time produced a homogeneous microstructure with higher crystallinity, lamellae thickness and crystalline − amorphous interfacial area, which led to remarkable increase in impact strength from 400 to ~ 1000 J/m and 6-fold increase in elongation from 2.5% to ~ 17% compared with the aged sample [43]. The effect of aging and annealing on PHB blends with three industrial plasticizers were subsequently investigated, including triethyl citrate (TEC), tri(ethylene glycol) bis(2-ethyl hexanoate)(TEG-EH) and tributyl citrate (TBC) [44]. The results showed an increase in impact strength for all the blends with plasticizers. However, both the elongation at break and tensile strength decreased due to cracks and voids emerging from contraction of the amorphous interlamellar layer in the matrix. The plasticizer was forced to leach out, which eventually led to deteriorating of properties instead of toughening effect [44].

Fig. 6. Proposed mechanism for the lamellar structure alteration after the annealing treatment.
Copyright (2014) Wiley Society of Chemical Industry [44].A recent investigation done by Hufenus et al. [45] focused on the effect of additives, various filament drawing setups and stress annealing on PHB fiber with an addition of commercial additives, boron nitride (BN) as a nucleating agent and tri-n-butyl (TBC) as a plasticizer to improve its melt-spinning. It was observed that crystallization was stabilized by the nucleating agents, and the damping viscosity fluctuations were achieved from plasticizers, thus, minimizing thermal degradation. On the other hand, the introduction of the modified draw-off unit enhanced the initial crystallization, thus, suppressing the post-crystallization [45]. The resulted drawn fibers crystallized in an orientation prior to a complete solidification, forming highly oriented amorphous phase (β-form) being kinetically confined between the aligned α-phase crystalline as observed by the wide-angle X-ray diffraction (WAXD). The fiber exhibited the highest tensile strength of 215 MPa at annealing condition 110 °C for 60 min, a 39% increase from neat fiber but with a decrease of 16% in tensile strain [45]. While annealing seems to be a remedial and effective method in the reduction of the brittleness of PHB-based materials and elimination subsequent aging of the samples, the main concern still lies in the compensation of toughness and elongation at break. In addition, the intricacy and high cost remain a challenge for up-scaling and adoption from the laboratory scale to industrial scale.
3. PHB toughening through blending
Blending is by far the easiest, most effective and economical approach for obtaining new materials with improved physical and mechanical properties, in which the drawbacks of the parent components can be altered and specially tailored by choosing and varying the compositions of the blend and preparation parameters. More importantly, blends of PHB with biodegradable polymershave been of much interest, in line with the concept of sustainability and eco-friendly. The concept alludes to the fact that the choice of materials, both matrix, and fillers, should be sourced from renewable resources. In this section, various blends of PHB with natural or synthetic sources and also non-biodegradable polymers are discussed.
3.1. Blending with materials from natural sources
3.1.1. Starch
Starch is an amylose-amylopectin storage polysaccharide as shown in Fig. 7, which is abundant, renewable and biodegradable. This bio-filler has found applications in textile industry, as sizing/stiffening agents and, as adhesives in papermaking. Blending of PHB with starch has been widely investigated with the sole aim of exploiting inherent properties of starch, in order to overcome PHB limitations and over the years, remarkable studies have been done on physical mixtures of starch and PHB.
 Fig. 7. Chemical structure of starch.
Fig. 7. Chemical structure of starch.For instance, in 2002, Godbole et al. [46] investigated the miscibility, morphology and thermomechanical properties of PHB/Starch blends, prepared through solvent casting technique. Thermal characterization of the blends revealed single glass transition temperature (Tg) in the range of 63 °C–87 °C, a strong suggestion that all the blends were miscible, in all the proportions tested [46]. On the other hand, DSC analysis exhibited crystalline nature of the blends, with melting peaks in the range of 165–168 °C. From the various proportions studied, an optimum tensile strength was reported at PHB/starch ratio of 7:3, an increase of about 72% compared to neat PHB's strength of 18.29 MPa [46]. It is worth noting that blending of 30 parts of inexpensive starch to 70 parts of costly PHB, results in substantial cost reduction. Additionally, these blends were thermally stable at about 203–223 °C, which was 30 °C lower compared to PHB/thermoplastic starch blends. The newly affordable bioplastics, with uncompromised properties, widens PHB applications landscape, in paper coating and cardboard packaging fields [46]. In a similar study, Zhang et al. [47]examined PHB properties by blending with dissimilar maize starch, one containing 70% amylase (starch 1) and, another containing 72% amylopectin(starch 2). In this work, similar PHB/starch ratio of 70:30 was employed and processed through melt compounding. The results revealed the dual action of starch, both as filler and nucleating agent in PHB/starch blends, which led to considerable reduction in the size of PHB spherulites [47]. As a result, thermomechanical properties were significantly enhanced, especially, for PHB and high amylose starch. From dynamical mechanical thermal analysis (DMTA), PHB/starch 1 blend exhibited a lower and narrower tan delta peak, compared to PHB/starch 2 blends. These results indicated that PHB/starch 1 blend, dissipated less energy due to strong interfacial bonding. This was ascribed to uninterrupted hydrogen bonding between PHB and high amylose starch, which is linear but coiled, compared to branched amylopectin starch (starch 2) [47]. In addition, the Tg of PHB/starch 1 was 2 °C lower than that of PHB/starch 2 (Tg ~ 37 °C), which was attributed to linear structure of amylose, compared to cross-linked amylopectin. Furthermore, PHB/starch 1 blends had higher thermal stability, as a result of PHB's carbonyl and starch's hydroxyl groups' interaction, which inhibited ring ester decomposition (chain scission) mechanism [47].
In an earlier work, Koller and Owen [48] studied the structure and mechanical properties of maize starch-filled PHB, as well as PHB/HV copolymer prepared by compression molding at 190 °C for PHB/starch. In this work, PHB/starch exhibited brittle failure at below 1% strain, indicating that starch caused PHB embrittlement, with increased modulus and lower strength [48]. In addition, PHB filled with destructured starch (treated in water under heat and shear) had better properties, especially, modulus compared to native starch granules. The effect was attributed to small particles size, asymmetrical shape and surface roughness of destructured starch [48]. Similarly, Thiré et al. [49] investigated the effect of starch on properties of compression molded PHB/starch blends at different starch contents, up to 50%. The results showed that thermal stability was not affected by the addition of starch, in all compositions. The onset degradation temperature was about 274 °C. On the other hand, elongation at break and tensile strength decreased with increased starch content, whereas, Young's modulus was unaffected up to 30% starch content [49]. Further, the modulus declined rapidly by about 70%, resulting in flexible materials, and this was as envisaged and, attributed to the ingrained rigidity of PHB. The overall poor mechanical properties of the blends were linked to inadequate interface adhesion and heterogeneous morphology of PHB/starch granules blends [49].
Innocentini-Mei and co-workers [50], explored the effects of chemical modification of starch on PHB's thermomechanical properties, by comparing natural starch, and modified starch i.e. starch adipate and urethane-grafted starch at varying proportions. In this study, it was discovered that natural starch and its derivatives, lowered the melting (Tm) and glass transition temperatures (Tg) in all the binary blends, compared to pure PHB [50]. However, much lower Tm and Tg values were observed for PHB/modified starch blends, ascribed to internal plasticization. On the other hand, all the PHB/natural starch and PHB/starch adipate blends were stiffer and brittle (low tensile strength at break), in spite of insignificant changes in elongation at break [50]. Unlike other studies, Lai et al. [51] discussed the possibility of enhancing properties of thermoplastic starch (TPS) by blending with PHB as a filler and TPS as a matrix. In this work, three kinds of gelatinized starch (i.e. potato starch, corn starch and soluble potato starch), were employed and blended with certain amounts of PHB, in order to tune the performance of TPS [51]. Significant changes in mechanical properties for the blends were noted. For instance, with a low degree (25% glycerol) gelatinized potato starch/7% PHB, the tensile strength increased 1100% and was ascribed to [1] high strength of PHB and, [2]reasonable low levels of polymer segregation in TPS matrix. On the contrary, 33% glycerol-TPS, exhibited poor strength, even at increasing PHB content, and this was attributed to high plasticizing effects [51]. Moreover, TPS (potato starch) had exhibited better mechanical properties improvement compared to TPS (soluble starch), which was linked to a higher molecular weight of potato starch and it's highly bonded cohesive structure. Additionally, inclusion of PHB into TPS conferred significant thermal stability to the blends, a great potential for processability of new environmentally benign plastics [51].
Besides the unmodified binary PHB/starch blends, researchers have further studied complex PHB/starch blend systems, including ternary blends. For instance, Ma et al. [52] investigated the physical properties of PHB/EVA/starch blends by reactive compatibilization using maleic anhydride (MA) and benzoyl peroxide (BPO). In this work, EVA/starch/MA/BPO/glycerol were pre-compounded, and then blended with PHB [52]. Fig. 8 shows SEM images gelanatized starch, uncompatibilized EVA/starch, compatibilized EVA-g-starch and proposed reaction mechanism for compatibilization. As shown, starch particles in compatibilized blend are homogeneously dispersed in EVA matrix, which was then introduced into PHB to produce PHB/EVA-g-starch blends [52].

Fig. 8. SEM images showing morphologies of gelatinized starch, EVA/starch (50/50 wt%) and EVA/starch (50/50 wt%) compatibilized with 0.09% MA.
Copyright (2014) Elsevier [52].The authors reported that tensile strength increased with MA content up to 0.09%, but later decreased due to change in phase morphology and size of starch particles and EVA domains. Additionally, the co-continuous morphology resulted in an increase in elongation at break and, hence improved toughness of the blends. The enhanced toughness was ascribed to the improved affinity of EVA-g-starch and PHB, compared to PHB/starch blend [52]. Cavitation, fibrillation and matrix yielding were identified as the major toughening mechanisms in reactively compatibilized PHB/EVA/starch blend system. On the other hand, thermal properties of PHB were somehow inert to reactive compatibilization process. However, the Tg of EVA component increased by 9 °C with 36% increase in MA content, attributed to miscibility and morphological changes, arising from composition array of EVA domains [52]. In another interesting study by Liao and Wu [53], PHB-g-acrylic acid (PHB-g-AA) was synthesized by grafting acrylic acid (AA) to PHB, initiated BPO, and subsequently melts blended with starch for comparison with PHB/starch blend, in terms of thermomechanical properties. The mechanical properties of PHB-g-acrylic/starch blend were found to be superior to PHB/starch blend up to 50% starch content [53]. The enhancement in properties was ascribed to the reaction between hydroxyl groups in starch and carbonyl group in PHB-g-AA and hence, improved compatibility in these blends compared to PHB/starch blends. In addition, PHB-g-AA/starch blends had improved processability compared to PHB/starch blends, due to low melt viscosity [53]. The authors reported that the melting temperatures (Tm) of all the blends decreased with increased starch content up to 50%, and were in the range of 170 °C–173 °C and 168 °C–172 °C, for PHB/starch and PHB-g-AA/starch, respectively [53].
Polyvinyl acetate (PVAc)-modified starch has also been synthesized and used to improve the thermal and mechanical properties of PHB. In a recent work, Don et al. [54] grafted vinyl acetate (VAc) to potato starch, initiated by ceric ion, to obtain PVAc-modified starch, which was subsequently blended with PHB. In this study, it was discovered that all PHB/PVAc-modified starch blends were miscible at all combinations [54]. This was ascertained by single glass transition temperatures (Tg) for the blends, in both DSC and DMA analysis. Furthermore, TGA results showed improvement in thermal stability with the addition of PVAc-modified starch, with a three-stage degradation profile, compared to single degradation pattern for PHB at 288 °C [54]. In essence, the improved thermal stability of PHB by PVAc-modified starch meant an enlarged processing window for PHB. In terms of mechanical properties, PHB/PVAc-modified starch blends were found superior in toughness, compared to virgin PHB. This was ascribed to improved compatibility and leathery nature of PVAc-modified starch [54].
3.1.2. Cellulose and its derivatives
Cellulose is the most abundant structural polysaccharide on earth, mainly found in plants. It consists of glucosidic linkages, where C-1 of one glucose unit joins C-4 of the next glucose molecule as shown in Fig. 9. On a nanoscale, it forms nanofibrils with unique strength properties that make up the structure of the plants. As a result of these unique properties and nanostructure form, cellulose nanocrystals (CNCs) have been widely investigated as bio-fillers for PHB polymer.
 Fig. 9. Chemical structure of cellulose.
Fig. 9. Chemical structure of cellulose.For example, Seoane et al. [55] thoroughly studied the effects of CNCs on PHB properties. In this study, DSC cooling scans demonstrated that CNCs were effective nucleating agents for PHB in all the nanocomposites, with an exothermic peak at ~ 83 °C, compared to ~ 62.5 °C for pristine PHB. This indicated that CNCs effectively reduced the energy barrier for PHB nuclei formation [55]. Moreover, CNCs were found to favor PHB crystallization, with 1.4% change of in crystallinity. Worth noting, pristine PHB exhibited single degradation pattern, with maximum DTG peak at ~ 295 °C, whereas, PHB/CNC nanocomposites had two-step profile, with lower decomposition temperatures compared to pristine PHB. This was attributed to the presence of sulfate and hydroxyl groups on CNC, which assisted chain scission of PHB at elevated temperatures [55]. On the other hand, with up to 6% CNC content, tensile modulus and strength of the nanocomposites increased by ~ 50% and ~ 35%, respectively. Furthermore, nanoindentation tests exhibited high elastic moduluswith CNCs up to 2% and hardness values in the range of 155–165 MPa, which suggested the effective reinforcing effect of CNCs and good CNC-PHB matrix interactions [55]. In a similar study, S de O Patrício, Patrícia, et al. [56] evaluated the influence of cellulose nanowhiskers (CNWs) on thermomechanical properties of PHB. In this research, CNWs dispersion in PHB matrix was facilitated by polyethylene glycol (PEG) by solution casting method. TGA results showed the enhanced thermal stability of the blends in two stages at 228 °C and 295 °C, corresponding to PEG and PHB degradation, respectively [56]. Additionally, melting temperatures of the blends were reduced by ~ 9%, compared to PHB. The TGA and DSC results indicated a widened processing window for PHB, by incorporating CNWs. In terms of mechanical properties, toughness (viz. strain at break) of the nanocomposites improved 50-fold with up to 0.45 wt% CNW and 15 wt% PEG, with strength remaining almost unchanged at 17 MPa. This was attributed to preferential chain orientation in the tensile direction. However, Young's modulus declined remarkably due to reduced resistance to chain flow, arising from capsulation of CNW by PEG. These materials potentially extend application possibilities for PHB [56].
In one recent study, Wei and co-workers [57] reported on in-situ compatibilized PHB-cellulose materials obtained from free radical polymerization, initiated by dicumyl peroxide (DCP). Fig. 10 shows the predicted schematic structures obtained through the generation of free radicals and grafting sites in PHB/cellulose. From this study, the authors found that grafting was dependent on time and DCP concentration [57]. Consequently, the thermal stability of PHB was enhanced, as a result of the formation of new bonds. Although mechanical properties were not reported, chemical cross-linking in PHB-cellulose materials could facilitate effective stress transfer. [57]

Fig. 10. Schematic illustration showing possible free radical sites and grafting sites in PHB/Cellulose blends initiated by DCP. Reprinted with permission from ref.
Copyright (2015) American Chemical Society [57].In another work, Zhijiang et al. [58] prepared PHB-bacterial cellulose (BC) biocompatible scaffolds by impregnation of BC into PHB-chloroform solution, and subsequent freeze-drying to remove the solvent. In this study, it was discovered that incorporation of BC, increased overall crystallinity of PHB in PHB/BC scaffolds. This suggested that BC nanofibrils have crystallization capability influence on PHB molecules. On the other hand, tensile strength and elongation at break, increased 13-fold and 2-fold, respectively [58]. This was attributed to reinforcing effects of BC in PHB matrix, arising from the inherent high strength of BC. Similarly, Barud and co-workers [59] also prepared BC/PHB membranes, with up to 90 wt% PHB by solution casting technique. The authors reported improvement in thermal stability of BC/PHB membranes with maximum temperature of ~ 360 °C, compared to ~ 320 °C for pure PHB [59]. In addition, above 50 wt% PHB, overall crystallinity was enhanced, although lower than pure PHB. Young's modulus, tensile strength and elongation at break were found to be dependent on BC/PHB combinations, and were reported to be as high 144 MPa, 9.5% and 15.5 GPa, respectively [59].
Interestingly, cellulose being a unique polymer with hydroxyl groups offers plenteous room for surface modification on the polysaccharide backbone, such as, etherification, esterification and so on. A number of cellulose derivativeshave been produced, including cellulosic derived plastics such as cellulose acetate (CA), ethyl cellulose, cellulose acetate butyrate (CAB), and cellulose acetate propionate (CAP). In recent years, these cellulose derivatives have become of particular interest to research groups in tuning the properties of biodegradable PHB polymer. For instance, Zhang and Deng [60] prepared biodegradable PHB/hydroxyethyl cellulose acetate (HECA) blends by solution casting method. Although the authors did not report on the mechanical properties, thermal changes of the blends were clearly observed. In this regard, melting temperatures of blends with PHB above 20% was about 175 °C, and were independent of the blend composition [60]. Low PHB content (˂20%) blends exhibited depressed equilibrium melting temperature of PHB, and this was attributed to mesophase-isotropic phase transition of HECA within the same temperature range. Furthermore, Tg of PHB in all the blend compositions remained unchanged at about 8 °C. In addition, HECA content in the blends markedly affected both nonisothermal and cold crystallization, with a shift to lower temperatures and higher temperatures, respectively [60]. Zhang et al. [61]also explored thermal properties of solution cast PHB/ethyl cellulose (EC) binary blends. In this study, increase in EC content in the blends caused an upward shift in the observed composition-dependent Tg. However, Tg of PHB remained unchanged at approximately 5–9 °C for all the blends. Both, the melting temperatures and crystallinities of PHB/EC blends were superior to that of pure PHB. Mechanical properties of these blends were not reported [61]. In a similar work, Chen and co-workers [62] investigated mechanical properties of PHB/EC blends prepared through melt mixing technique. In this work, PHB/EC blend system was reported to be heterogeneous; in which the dispersed EC domains acted as melt viscosity modifiers in processing and, as stiffeners under shear flow [62]. The dual character of EC domains influenced the spherulites structure of PHB, as well as its kinetics of crystallization. For example, inclusion of EC led to (i) reduced growth rate of PHB spherulites, (ii) reduced crystallinity and (iii) lamellar defects, which subsequently, affected the mechanical properties of PHB. With low EC (˂1 wt%), EC acted both as reinforcing and toughening agent. The impact and tensile strength of blends with 1 wt% EC were enhanced by 1.22 and 1.15 times, respectively [62].
Yamaguchi and Arakawa [63] investigated the structure-properties relationships of PHB with four different kinds of cellulose derivatives (i.e. CAP and CAB), including the mechanical properties. The blends were prepared by mixing and compression molding at 180 °C, followed by crystallization at 40 °C for 5 min. In this work, the authors reported anomalous viscoelastic behavior of the blends observed through DMA. Although, high molecular weight cellulose derivatives CAP46 and CAB37 were brittle, their blends with PHB showed unlikely plastic deformation [63]. This was attributed to reduced crystallinity of PHB, as a result of more entrapped floating chains. These floating chains resulted to more amorphous PHB blends, thus, ductile failure mode under DMA and tensile tests was reported. These results agreed with the shift in β-relaxation peaks to lower temperature [63]. On the other hand, PHB blend with low molecular weight CAB52 and CAB38 exhibited low elongation at break (˂1%) and comparable β-relaxation peak as PHB and PHB-rich blends at about 20 °C. In conclusion, mechanical properties of PHB/cellulose derivatives blends can be tailored by fine-tuning the amorphous fraction [63].
3.1.3. Lignocellulosic
Plant matter (biomass) is the most abundant resource on earth, widely used in biorefineries. The biomass is composed of cellulose, hemicelluloses and lignin, which have important roles on overall structural properties of lignocellulosic materials [64], [65]. In light of this, value addition of plant matter, as additives in polymers, through valorization is advantageous. In an earlier study, Angelini and coworkers [66] fabricated PHB blends with crude lignin-rich residues (LRR) and alkaline lignin (AL) through compression molding. In this work, LRR had no significant effect on thermal stability of PHB, whereas, AL acted as a strong pro-degrading agent. As anticipated, TGA analysis revealed that LRR had higher mass loss and lower char yield, which was due to decomposition of cellulose fraction present [66]. Moreover, the Tg values of AL and LRR were 112 and 113 °C, respectively. Tensile and impact tests showed overall decrease in mechanical properties of PHB/LRR blends. For example, PHB/30% LRR blend showed a decrease of about 60% and 55% in tensile strength and impact strength, respectively. This was ascribed to presence of defect sites between the phases, filler heterogeneity, poor compatibility and weak filler/matrix interface adhesion. On the other hand, flexural modulus of PHB/30% LRR blend was about 40% higher than that of pure PHB. This was ascribed to flexural modulus being dependent on the combined action of individual components in the blend and only slightly sensitive to components' mutual interfacial interactions [66].
The same research group [67] further investigated the effects of isolated fractions of lignocellulosic (LC) biomass on PHB properties. In this work, the authors obtained two fractions i.e. insoluble lignin (IL) and hollocellulose (HC), from biorefinery waste and used it as fillers in PHB matrix, in which purified LC was the reference [67]. LC 30% w/w was found to have remarkable enhancement on PHB's moduli and complex viscosity, which was almost independent of time and frequency. These results were supported by SEM observations, showing good dispersion of LC in the matrix that resulted in a tightened co-continuous network, with good filler-matrix interface. However, IL did not promote PHB crystallization compared to HC. This was ascribed to the higher melt viscosity of PHB/LC blends, which hampered the chain mobility [67].
Mousavioun et al. [68] investigated the thermal and rheological properties of PHB/lignin blends in different compositions. The authors reported 50% drop in thermal decomposition activation energy of PHB/lignin blends compared to pure PHB, which suggested that presence of lignin reduced the thermal stability of the blends. With up to 40% lignin content, the blends exhibited single phase morphology, single Tg (15 °C) and miscibility [68]. However, above 50% lignin content, phase separation occurred, with two Tg (18 °C and 130 °C) of the components. On the other hand, viscoelastic properties were dependent on lignin content. At low lignin content (≤ 30%), dynamic and loss modulus were lower than that of pure PHB, whereas, higher loadings of lignin exhibited higher moduli. These viscoelastic response differences between low lignin and high lignin content PHB blends were attributed to lower crystallinity and enhanced rigidity, respectively [68].
3.1.4. Chitin
Chitin is the second most ubiquitous structural polysaccharide after cellulose, found in exoskeletons of crabs, lobster, beetles, shrimps, as well as cell walls of fungi and yeasts. Fig. 11 show structures of (a) chitin and (b) chitosan. Chitin consists of both, crystalline and amorphous domains; the crystalline domains can potentially serve as reinforcements. Furthermore, chitin biofiller has many desired properties, including, biocompatibility, biodegradability, nontoxicity and high modulus. Upon hydrolysis, chitin yields chitosan.
 Fig. 11. Chemical structure of (a) chitin and (b) chitosan.
Fig. 11. Chemical structure of (a) chitin and (b) chitosan.Research studies have shown that introduction of chitin/chitosan as rigid filler into PHB matrix improved the mechanical properties. However, publications in this area of research are limited, probably due to (i) high cost of the common solvent for PHB/chitin/chitosan in case of solvent casting and, (ii) susceptibility of chitin/chitosan to carbonization under high temperature in melt blending [69]. However, prior arts in this area are worth highlighting. For instance, Cao and colleagues prepared PHB/chitosan films and scaffolds using emulsion blending technique, in which PHB in chloroform and chitosan in acetic acid were mixed, followed by casting/evaporation processes. Compared to neat chitosan films, 30% PHB/chitosan blend exhibited higher tensile strength and elongation at break, by an increment of 38% and 63%, respectively. Furthermore, these properties coupled with the porous structure of the PHB/chitosan films accentuate tissue engineering application possibilities for these composites [69]. In a particular study, Ikejima et al. [70] investigated thermal properties and crystallization behavior of PHB/α-chitin and PHB/chitosan. In this work, composites were prepared by solution casting in hexafluoroisopropyl alcohol (HFIP) solvent. PHB, chitin and chitosan solutions were prepared separately, admixed, cast on poly(tetrafluoroethylene) (PTFE) dish, dried at RT and under vacuum for a couple of days [70]. From DSC characterization, the authors revealed that both α-chitin and chitosan hampered PHB's crystallization. This observation was ascribed to the reduced PHB's lamellar thickness, intermolecular filler/matrix interactions, resulting in confined and trapped PHB molecules in the blends. Compared to α-chitin, chitosan had about 10% more suppressing power, obtained from the heat of fusion (ΔH) of varying blend compositions [70]. In another recent study, Khasanah et al. [71] prepared PHB/Chitin blends through solution casting technique. In this work, the authors explored the effects of intermolecular hydrogen bonding between carbonyl groups of PHB and amine groups of chitin. From the DSC cooling scans, the crystallization temperature of PHB was depressed at low chitin loadings (≤ 10 wt%), an indication of increased crystallization [71]. This improvement was linked to nucleating effect of chitin, which promoted rapid growth of PHB crystals. However, at higher chitin loadings, crystallinity decreased and, this ascribed to reduced PHB chain mobility, due to intermolecular hydrogen bonds between PHB and chitin. Fig. 12 shows the proposed structural changes and intermolecular hydrogen bonds interactions in PHB/Chitin blends [71].

Fig. 12. Proposed carbonyl-amine hydrogen bonds interactions in PHB/Chitin blends and the resultant structures.
Reprinted with permission from ref. Copyright (2015) Elsevier [71]