1. Introduction
Traditionally, pyrometallurgical processes have dominated metal extraction from ores, especially the sulphidic type. However, these processes are electrical energy intensive and as such, their application is more suited for high-grade ores as their use on low-grade ores is uneconomical. Furthermore, the ongoing power disruptions in South Africa pose a serious threat to the world’s leading producer of platinum group metals (PGMs). In contrast, hydrometallurgical processes can economically beneficiate high and low-grade and also complex ores.
Although hydrometallurgy has replaced pyrometallurgy in the extraction of many metals, there are still some metals like the PGMs that are not currently extracted industrially without some pyrometallurgical pre-treatment steps. PGMs are a family of six structurally and chemically similar elements that are most valued for their wide range of industrial, medical, and electronic applications (Glaister and Mudd, 2010). They are: Pt, Pd, Rd, Ru, Ir and Os. Together with silver and gold, PGMs are classified as noble metals due to their high oxidation potential and corrosion resistance (Xiao and Laplante, 2004, Glaister and Mudd, 2010).
Pyrometallurgy requires large volumes of high-grade feed for economic processing. The depletion of high-grade ores, therefore, implies decreased feed rates to the pyrometallurgical stages and consequently decreased productivity and profitability. The production of PGMs is primarily controlled by Southern Africa (South Africa and Zimbabwe) and Russia. However, due to the Ukraine war, there is a potential risk to Russian PGM sales. Additionally, in South Africa, frequent electricity load-shedding poses a significant threat to the electrical PGM smelting process. Consequently, this situation serves as a crucial catalyst for exploring alternative options that are less reliant on electricity consumption. Furthermore, with the ever-fluctuating metal prices, the profitability of metal processing is compromised even further. Thus, partial or total replacement with a low-cost hydrometallurgical alternative is imperative. To further improve the environmental friendliness of hydrometallurgy, bio-hydrometallurgy has attracted a lot of interest due to its inherent advantage of using naturally occurring microorganisms to produce lixiviants, with less environmental concerns associated with production and handling. Moreover, the process has been noted to have lower energy consumption (Bosecker, 1997; Rawlings, 2005). Furthermore, the re-use of microorganisms ensures a constant supply of the required lixiviant (Ndlovu, 2008).
Considering the economic and environmental advantages of bio-hydrometallurgy, it would be beneficial to expand the application of this technology in the processing of PGMs (Mwase and Petersen, 2017) (Mpinga et al., 2018). An additional incentive to explore the bioprocessing of PGMs stems from the imminent depletion of high-grade ores that yield concentrates suitable for smelting. The current approach involves blending Merensky Reef ores with UG2 ores to address smelting challenges.
The Merensky Reef is exceptionally rich in platinum group elements, occurring in the form of sulphide minerals like cooperate and braggite (Cramer, 2001). It also contains chromite and sulphides. In terms of conventional PGM processing methods, the Merensky Reef is the most economically viable option (Cramer, 2001). For instance, the Merensky Reef is ten times more abundant in base metal sulphides (BMS) than the UG2 Reef (Solomon et al., 2011). However, as the Merensky Reef is being depleted, it becomes crucial to utilize other PGM reserves.
The Platreef also holds a significant amount of economically viable PGMs, albeit at a lower grade compared to the Merensky and UG2 Reefs. The mineralogy of the Platreef is highly complex, with sperrylite being the most common PGM mineral (Cramer, 2001). The Platreef also contains substantial quantities of pyrrhotite (Cramer, 2001). On the other hand, the UG2 Reef primarily consists of chromite, with PGMs occurring as sperrylite, braggite, and cooperate (Mpinga et al., 2018). In the UG2, low-grade PGM concentrates are sometimes associated with high chromite contents. The high pyrrhotite content in Platreef ores and the elevated chromite levels in UG2 ores pose challenges to smelter efficiency (Mwase et al., 2012a).
In some cases, PGMS are locked in silicates and report to the tailings. For example, it has been reported that almost 50% of PGMs in the Platreef ores deport to silicate minerals (Bryson, 2008) resulting in a significant fraction of PGMs being lost to the tailings. There is, therefore, a need to also look into silicate-destroying microorganisms when developing a bioleaching process for PGMs. In other cases, the reefs are associated with mineral grade inconsistencies, meaning that some regions might have very low PGM grades that are not suitable for the concentrator-smelter-refinery route. Grade inconsistency has also been reported for the Platreef ores (Mwase et al., 2012b). All the challenges mentioned above suggest that there is a significant need to look at other PGM processing alternatives.
Conventional leaching of the platinum group metals has been through use of chlorides (Tuncuk et. al., 2012) or cyanide (Hourn and Turner, 2012). Thus, if a bioprocessing route would be developed, it follows then that chlorine or cyanide-producing microorganisms might be a good option. However, whereas cyanogenic microorganisms have been identified and isolated, chlorine-producing microorganisms have not yet been identified. Therefore, the use of cyanide producing microorganisms is the best starting point in developing a process for the bioleaching of PGMs.
The importance of recovering PGMs is evident in the circular model of metal resource recovery from urban wastes. Due to their high value and depletion of deposits, PGMs are among the metals that have been targeted for metal recycling. The recycling of PGMs may be done by conventional chemical recovery methods or new technologies such as biotechnology (Dodson, 2015; Karim and Ting, 2021). The low-cost requirements and environmental friendliness of biotechnology make this option attractive (Liang and Gadd, 2017). However, the aim of this paper is to provide the necessary knowledge for the bioleaching of PGMs from primary sources. The sections that follow provide a background of the mineralogy of PGM ores and their conventional processing, a description of the bioleaching of the major mineral phases associated with PGMs, the challenges and opportunities of PGM bioleaching and a suggested processing route for the bioprocessing of PGM ores.
2. PGM occurrence
Proterozoic and Archaean age layered intrusions (e.g. the Bushveld Complex, South Africa and the Great Dyke, Zimbabwe) are the world’s foremost sources of PGMs (Oberthür et al., 2018). Over 80% of the earths reserves are contained in the Bushveld Complex of South Africa (Junge et al., 2015). Generally, PGMs are associated with either chromitites or sulfide-rich rocks (Scoon and Teigler, 1994, Naldrett and Naldrett, 2004). In chromite environments, PGMs occur in alloys, sulphides, arsenides, sulpharsenides (Junge et al., 2014; 2016; Barnes et al., 2016). Bismuthotellurides are generally found in base metal sulphide systems such as the Merensky Reef, the Great Dyke and the Platreef (Kinloch, 1982, Oberthür et al., 2003, Holwell and McDonald, 2007, Osbahr et al., 2013, Junge et al., 2015). PGM sulphides and arsenides are also found in base metal sulphide systems (Junge et al., 2019).
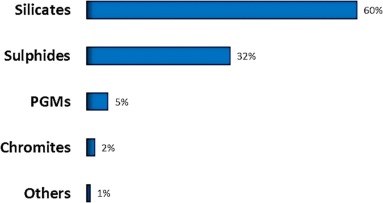
Although, silicates are a high constituent in PGM ores (Fig. 1), and sometimes contain most PGMs, generally PGMs are associated with the sulphide phases, as reported in previous work (Seymour and O'Farrelly, 2001, Schouwstra et al., 2000). They exist within base metal sulphide (BMS) minerals or at BM-silicate grain boundaries. For this reason, milled PGM ores can be floated to recover PGMs (Junge et al., 2019). Generally, PGMs are associated with the BMS (Lee, 1996). Virgin ores consist mainly of pyrrhotite (Fe1-xS) (∼75%), pentlandite (Fe, Ni)9S8), chalcopyrite (CuFeS2) and minor quantities of pyrite (FeS2) (Junge et al., 2019).
 Fig. 1. Mineral association proportions based on MLA data for a PGM concentrate from the Bushveld Complex of South Africa.
Fig. 1. Mineral association proportions based on MLA data for a PGM concentrate from the Bushveld Complex of South Africa.Some PGMs are also found in oxides that form as weathering products of sulphide ores (Oberthür et al., 2013). During weathering of sulphide ores, there is a redistribution of PGMs and a substitution of base metal sulphides by iron oxides or hydroxides (Junge et al., 2019). The base metal sulphides are oxidised when there is low fugacity of sulphur (Augé and Legendre, 1992). As can be seen in Fig. 1 (based on Junge et al., 2016), the most common non-sulphide phases in PGM ores are silicates and chromites.
3. Conventional processing of PGMs
Conventional processing of PGM ores consists of mineral beneficiation (flotation), pyrometallurgical processing (matte smelting) and hydrometallurgical extraction (leaching) (Crundwell et al., 2011). A typical flowsheet showing the main processes involved in the processing of PGMs is shown in Fig. 2 (based on Cramer, 2008).
 Fig. 2. Conventional Processing of PGMs.
Fig. 2. Conventional Processing of PGMs.Smelting sub-processes include concentrate drying, electric arc smelting to remove slags, converting of matte to reduce sulphur and iron, gas cleaning, production of H2SO4 from SO2, matte granulation and crushing of matte. Base metal refining consists of sub-processes such as sulphate leaching, impurity removal, copper electrowinning, cobalt removal and recovery, nickel electrowinning or hydrogen reduction and sulphuric acid production from SO2. PGM separation and refining consists of sub-processes such as leaching, solvent extraction, ion exchange, precipitation, distillation, effluent treatment and metal preparation for dispatch.
4. Direct leaching of PGMs
Several hydrometallurgical approaches have been proposed for the extraction of PGMs (Mpinga et al., 2015). The complex PGM mineralogy has made the development of a universally feasible process difficult. Of relevance to this paper are those processes that use cyanide for the extraction of PGMs as it is anticipated that leaching with biogenic cyanide will require more or less the same parameters as those for inorganic cyanide leaching.
Hydrometallurgical extraction of PGMs is motivated by the need to profitably process low-grade and mineralogically complex ores within strict environmental regulations. Conventional smelter-based operations are only applicable to high-grade, low-chromite, large resources and long life-of-mine operations (Mpinga et al., 2015). Several direct leaching approaches to PGM extraction have been proposed, but of interest to this paper are the commercial applications that use the cyanide lixiviant (Albion process (Fig. 3) and the pressure oxidation processes (Fig. 4)). Several authors have also looked at direct PGM extraction techniques from secondary materials, especially spent automotive catalyst. Table 1 provides additional pre-treatment steps that have yielded improved PGM recoveries.
 Fig. 3. Albion process for the hydrometallurgical extraction of PGMs (Mpinga et al., 2015).
Fig. 3. Albion process for the hydrometallurgical extraction of PGMs (Mpinga et al., 2015). Fig. 4. Total Pressure Oxidation process for the hydrometallurgical extraction of PGMs (Mpinga et al., 2015).
Fig. 4. Total Pressure Oxidation process for the hydrometallurgical extraction of PGMs (Mpinga et al., 2015).Table 1. The extraction techniques employed for the pre-treatment and subsequent recovery of precious metals (PGMs) from secondary sources.
| Material used – and metals extracted | Pre-treatment method | Highlights | References |
|---|---|---|---|
| Spent automotive catalyst – Pt, Pd and Rh | Ultrasound assisted nitric acid | 80% ultrasonic power, 37 kHz ultrasonic frequency, 6 M HNO3, 50 min. Accelerated leaching, improved recoveries, reduced reagent consumption, lowering temperature requirements. Recoveries of 38% Pt, 44% Pd and 91% Rh were achieved. | Karim and Ting (2022) |
| Spent automotive catalyst – Rh | Oxidation followed by reduction | Oxygenation followed by addition of hydrogen. Resulted in Increased dissolution rates. Recoveries of 56% Rh were attained. | Chen et al. (2014) |
| Spent automotive catalyst – Pt, Pd, Rh | Use of formic acid | PGMs were leached with electro-generated chlorine after pretreatment of spent automotive catalyst with formic acid. High extractions are achieved at low concentrations of formic acid. Recoveries of 97% Pt, 94% Pd and 90% Rh were attained. | Upadhyay et al. (2013) |
| Spent automotive catalyst – Pt, Pd, Rh | Zn vapour deposition | Zn vapour treatment of spent automotive catalytic converters prior to acid leaching. Resulted in improved recoveries of 98% Pt, 97% Pd and 65% Rh. | Sasaki and Maeda (2014) |
| Spent automotive catalyst – Pt | Coke oxidation | Burning of the carbonaceous material supporting the catalysts. Resulted in 100% Pt solubilisation. | de Sá Pinheiro et al. (2004) |
| Na2PtO3 – Pt | Calcination | Formation of a (Na,Li)2PtO3 solid solution by heating Pt in the presence of Li2CO3 at 800 °C. Ability to dissolve in relatively safe lixiviants such as HCl. Resulted in 96 – 97% Pt recovery. | Kasuya et al. (2014) |
| Spent automotive catalyst – Pt, Pd, Rh | Mechanochemical activation |
Done in the presence of an oxidising agent – details still concealed. Resulted in 77.2% Pt, 97.4% Pd and 62.1% Rh. |
Wei et al. (2019) |
5. Postulating the bioleaching of PGMs
5.1. First step - bioleaching of base metal sulphides associated with PGMs (Pretreatment of PGM concentrates)
The removal of base metals prior to the PGM extraction process is important in order to minimise their interference during the PGM leaching stage. Base metal removal can be done using mesophilic and thermophilic microorganisms. With respect to base metal leaching from PGMs, previous work (Mwase et al., 2012a, Mwase et al., 2012b, Mwase et al., 2014, Eksteen et al., 2014) on Platreef PGM concentrates, used columns of granite coated with slurry and a mixture of thermophiles and mesophiles. Recoveries of 91.1% Cu, 98.5% Ni and 83.5% Co were achieved after 88 days of leaching at 65 °C. The setup for the leaching tests is shown in Fig. 5 (Mwase et al., 2012a).
 Fig. 5. Schematic layout of the It can be deduced from this paper that from PGMs (Mwase et al., 2012a).
Fig. 5. Schematic layout of the It can be deduced from this paper that from PGMs (Mwase et al., 2012a).After successful removal of base metals from PGM concentrates, the next stage is the leaching of PGMs. However, since, as reported earlier, some PGMs report to silicates, and get lost to the tailings, it would be important to biochemically decompose the silicate minerals, prior to the extraction of PGMs.
5.2. Second step – Decomposition of silicates
The advantages of using microorganisms, instead of chemicals, for silicate decomposition include environmental friendliness, cost-effectiveness and high selectivity. Silicate-decomposing microorganisms are known to be much more environmentally gentle compared to toxic chemicals, resulting in significantly reduced environmental impact (Dwyer et al., 2012). Moreover, employing silicate-decomposing microorganisms instead of chemicals offers overall cost-effectiveness since microorganisms can be cultivated using low-cost substrates such as agricultural waste. Other studies (Wonyen et al., 2018, Faramarzpour et al., 2022) have reported improved selectivity and enhanced process efficiency with the use of microorganisms, and the same is anticipated with the use of silicate-decomposing microorganisms. The effectiveness of microorganisms for silicate decomposition can vary depending on the specific mineral composition, pH, temperature, and other environmental factors (Lauwers and Heinen, 1974). Therefore, there is a need to explore various experimental options for the use of microorganisms for silicate decomposition in PGM ores.
According to Henne and co-researchers (2018), A. ferrooxidans are able to solubilise some Fe2+-bearing silicates such as chlorite and amphiboles that are also associated with PGMs. It is, therefore, likely that some degree of silicate decomposition will take place during the bioleaching of PGMs with either mesophilic or thermophilic microorganisms. Henne and co-researchers (2018) found that the dissolution of silicates provided access to sulphides trapped within the silicates. Earlier work by Welch and Ulman (1996) also revealed the possibility of solubilising a number of silicates using A. ferrooxidans and F. acidarmanus.
Dopson and co-workers (2009) were also able to solubilise silicates using F. acidarmanus and A. ferrooxidans. The authors reported that the microorganisms tend to grow on the dissolution of the silicate minerals. They found that during the dissolution of the silicate mineral olivine, there was a release of Fe2+ that might have become a substrate for the microorganisms resulting in increased leaching rates. The finding by Henne and co-workers (2018) that A. ferrooxidanstend to attack Fe2+ silicates is consistent with those by Dopson and co-researchers (2009). However, since some major silicates associated with PGMs such as serpentine and are not iron-bearing, these might not be amenable to dissolution with A. ferrooxidans. The tendency of silicate minerals to dissolve in the presence of acidophilic microorganisms has also been reported by several other researchers (Bhatti et al., 1993, Bhatti et al., 1994, Tasa et al., 1995, Bigham et al., 2001, Santelli et al., 2001, Welch and Banfield, 2002). Using a mixed culture of acidophilic iron- and sulphur-oxidising bacteria, Bhatti and collaborators (1993) bioleached mixed silicates that were mostly phlogopitewhereas Bigham and co-workers (2001) were able to solubilise mica. Using similar microorganisms, Bhatti and collaborators (1994) and Tasa and co-workers (1995) solubilised mixed silicates whereas Santelli and co-researchers (2001), Welch and Banfield (2002) targeted the silicate fayalite.
An indirect mechanism was proposed for the dissolution of silicates by bacteria (Avakyan et al., 1985). Avakyan and co-researchers (1985) dissolved silicon using the bacteria Sarcina ureae and observed the production of ammonia that in turn resulted in an alkaline medium which then disrupted the siliceous bonds. The formation of ammonia by Sarcina ureae is as shown in Equation (1)(Cook, 2017).(1)
Avakyan and co-researchers (1985) also found a similar result with the bacteria Paenibacillus mucilaginosus which was able to release silicon from quartz. The dissolution of silicon by Paenibacillus mucilaginosus was later investigated by Liu and co-workers (2006). They found that two processes were responsible for the decomposition of silicate minerals, i.e., the production of organic acids and polysaccharides by bacteria, followed by the adsorption of organic acids and SiO2 by the polysaccharides. The two processes altered the equilibrium between the mineral and liquid phases, and this caused the solubilising of silicon.
In addition to bacteria, it has been proven that fungal species such as Aspergillusand Penicillium are able to dissolve silicate minerals (Castro et al., 2000). Castro and collaborators (2000) found better extractions with A. Niger than with bacteria. They attributed the improved leaching kinetics of A. Niger to the production of citric acid by the fungal species. The citric acid produced by the fungal species results in the formation of complexes with metals (Castro et al., 2000). The complexation of metals in turn results in the rearrangement of the mineral structure and the consequent release of silicon (Ehrlich and Rossi, 1990). Other studies have also reported complexation in the decomposition of silicates by bacteria (Webley et al., 1963, Henderson and Duff, 1963).
However, the use of silicate-decomposing microorganisms also presents some disadvantages. These include challenges associated with process optimization and longer processing times compared to chemical methods. Microorganisms are highly sensitive to environmental changes, which can make it challenging to consistently maintain their efficacy (Dwyer et al., 2012). Achieving optimal conditions for microbial activity, such as strain selection, growth conditions, pH, temperature, and nutrient availability, requires careful monitoring and optimization. Additionally, the slower kinetics of microbial silicate decomposition can contribute to longer processing times compared to chemical-based methods. These factors highlight the need for ongoing research and development to address these challenges and improve the efficiency of using silicate-decomposing microorganisms in mineral processing.
It is also known that the dissolution of silicates is likely to influence conditions such as pH and viscosity, and might result in the production of fluoride ions, associated with the silicates, that might inhibit the further dissolution of other mineral sulphides (Dopson and collaborators, 2008). Dopson et al., 2009 also reported on the release of toxic fluoride elements during silicate decomposition. The thickening of leach liquor by silica was reported by Pietrobon and co-workers (1997). Brierley (2001) and Salo-Zieman and co-researchers (2006) reported the passivation of sulphide minerals by jarositeduring silicate dissolution. The formation of jarosite might be due to the low pH created during the oxidation of sulphide species associated with silicates and the dissolution of the iron associated with the silicates (Dopson et al., 2008). Given the challenges associated with silicate dissolution, the decomposition of silicates may be done as a separate stage, since the pH for silicate dissolution (3 to 5) (Henne et al., 2018) is higher than the pH for base metal leaching (∼1.5). Furthermore, it is known that acidophilic microorganisms preferentially attack sulphide minerals over silicate minerals (Edwards et al., 2000), perhaps due to the easier release of Fe2+ from the iron-sulphide bond.
It is anticipated that after the decomposition of silicates, the PGMs will be more amenable to bioleaching. The next section looks into the bioleaching of PGMs following the pretreatment stages. A background of PGM cyanide leaching chemistry is presented followed by a closer look into the possible approaches for the bioleaching of PGMs.
5.3. Third step – Bioleaching of PGMs
5.3.1. PGM cyanide leaching chemistry
Almost all transition metals form complexes in the presence of cyanide (Marsden and House, 2006). These complexes show high water solubility and chemical stability. It can be deduced that the reactions are similar to those of gold. However, the thermodynamic aspects of PGMs are different from those of gold, being associated with much higher oxidation potentials (Dorin and Woods, 1991, Marsden and House, 2006). Leaching of PGMs is not possible at room temperature and pressure due to poor kinetics (Chen and Huang, 2006). It is also known that the PGMs, namely Pd, Pt and Rh leach to different extents during pressure cyanidation, with Pd having the highest extraction efficiency and Rh having the least (Chen and Huang, 2006). Huang and collaborators (2006) also reported that the chemical stabilities of the PGM-CN complexes at high temperature were in the order Rh > Pt > Pd. The higher chemical stability of than is ascribed to different geometrical structures, with having an octahedral structure and having a planar structure (Huang et al.., 2006). The octahedral structure for implies the need to firstly break Rh-CN bond before oxidation (Huang et al.., 2006), unlike the planar structure that would not require breakdown of any bonds before oxidation (Huang et al.., 2006). The higher chemical stability for compared to is due to being heavier, and therefore being more thermodynamically and kinetically more stable (Huang et al.., 2006). Marsden and House (2006) and Mountain and Wood (1988) gave the stability constants (Log β4) for and as 78 and 63, and both were higher than that for (56). The Eh-pH diagrams for Pd and Pt (Dorin and Woods, 1991) reveal wide stability regions for both the Pd and Pt cyanide complexes. The metal/metal-CN complexes are pH dependent up to a pH of 9.2, beyond which they become independent of pH (Dorin and Woods, 1991). It can also be deduced that the formation of complexes will require oxygenated cyanide solutions, since the metal complexes exist below the oxygen line. The thermodynamic driving force for the formation of is greater than that for since there is greater difference in potential between formation and oxygen reduction reaction (Chen and Huang, 2006).
Mclnnes and co-researchers (1994) worked on ground ores (80% −74 μm) containing Pt, Pd and Au, and they observed the best recoveries for Pd (85%) at pH of 9.5 and at ambient conditions using NaCN lixiviant. However, Pt could not be extracted beyond 20% at the same ambient conditions. From the findings, it was observed that Au recoveries were higher than for both Pd and Pt, due to the weaker metallic bonding strength associated with Au (Huang et al.., 2006). Earlier work by Bruckard and co-workers (1992) realized 88 – 92% Pd recoveries and 73 – 79% Pt recoveries at 100 °C, pH of 9.5 – 11.5 for 4 – 6 h using atmospheric air. They reported that optimum leaching temperatures were between 100 and 125 °C. It is quite evident that high temperatures (>100 °C) are necessary for efficient PGM extraction. Huang et al. (2006), firstly removed base metals using acidic pressure leaching and then subjected the residue to pressure cyanidation for selective dissolution of PGMs, and subsequent zinc cementation to produce a Pt-Pd concentrate. Zinc cementation was done at 60 – 80 °C, pH 9.5 – 10, 2 h and 1 atm. Recoveries of > 95% and > 99% were attained for Pt and Pd, respectively, after the leaching stages. The leaching conditions by Huang et al. (2006) are summarised in Table 2.
Table 2. Leaching conditions for a PGM-bearing sulphide concentrate.
| Stage | Temperature (°C) | Lixiviant concentration (g/l) | Time (h) | O2pressure (MPa) | L:S ratio | Recoveries |
|---|---|---|---|---|---|---|
| Pressure acid leach (with H2SO4) | 200 | 12.5 | 6 | 1.8 | 4:1 | >99% Cu, Ni and Co |
| Two-stage cyanidation (with NaCN) | 160 | 6.25 | 1 | 1.5 | 4:1 | >95% precious metals |
Mwase et al., (2012a) used a laboratory scale column to investigate heap leaching of residual PGM concentrate following base metal removal with a mixed culture of thermophiles and mesophiles. They observed that some Rh and Pd, and to a much less extent Pt, were co-extracted with base metals. Dorfling et al., (2011) also reported similar co-extractions for sulphate based PGM refining. The subsequent cyanide leaching column tests were done at 23 °C (room temperature), and after 21 days, recoveries of 20.3% Pt, 87% Pd and 46% Rh were attained.
For the heap leaching of residual PGM concentrate, Mwase et al., 2012a deduced that Pd leached the most, with very little extractions for Pt and Ru. It can also be deduced from the figure that for both Pt and Rh, there was still leaching taking place when the tests were ended. Mwase and co-researchers (2012a) also inferred that during cyanide leaching of PGMs, not much of the gangue elements are leached. The relatively lower recoveries of Pt were attributed to its deportment to cyanide insoluble sperrylite. After increasing the leaching temperature to 50 °C and the leaching period to 45 days, Mwase and co-workers (2012b) were able to increase PGM recoveries. For example, the recovery of Pd increased to 96.5% and Pt recovery increased to 35%. After 60 days, recoveries of Pt and Pd were increased to 57.8% and 99.7%, respectively (Mwase et al., 2014). The cyanide leaching of PGM therefore appears to follow first order kinetics, and higher temperatures (>100 °C) should be expected to yield good recoveries for all PGMs.
Since NaCN leaching of PGMs has been found to be feasible, the use of cyanide-generating bacteria may result in an even more environmentally friendly process. The most common cyanogenic bacterial species are C. violaceum, Pseudomonas fluorescens, Pseudomonas plecoglossicida, Pseudomonas aureofaciens, Pseudomonas putida, Pseudomonas syringae, Pseudomonas aeruginosa, Bacillus megaterium, archaea Ferroplasma acidarmanus and Ferroplasma acidipholum, and some fungal species like Clitocybe sp, Marasmius oreades, and Polysporus sp. (Knowles, 1976, Askeland and Morrison, 1983, Paterson, 1990, Faramarzi et al., 2004, Faramarzi and Brandl, 2006, Hol et al., 2011). It is worth noting that several cyanide-based alternatives have been proposed for the leaching of PGMs. Shaik and Petersen (2017) conducted a comparison of SCN− + CN− + [Fe(CN)6]3-, SCN− + CN−, and SCN− + [Fe(CN)6]3- and found that SCN− + CN− + [Fe(CN)6]3- exhibited the highest effectiveness. These findings could be valuable for enhancing the bioleaching of PGMs using biogenic cyanide.
5.3.2. Production of biogenic cyanide
A large number of bacteria and fungi are able to produce cyanide (Møller and Seigler, 1999) and cyanide-producing microorganisms such as C. violaceum have been used for the bioleaching of gold from pre-oxidised refractory mineral ores(Lawson et al.., 1999). This is largely based on the historical effective and economic application of cyanide in the leaching of gold ores. Cyanide producing microorganisms such as C. violaceum that have been used for the bio-heap leaching of gold from low grade ores (Lawson et al., 1999), are therefore potential candidates for the bioleaching of PGMs since it is known that the gold and PGMs react with cyanide in a similar way (McInnes et al., 1994). A study conducted by Mwase and co-researchers (2012 a and b) proposed a heap chemical cyanidation process of PGM’s from low grade concentrates. This paper will therefore focus on bio-cyanidation of PGMs.
It is known that cyanide production only happens under microaerophilic conditions (Clawson and Young, 1913). The production of cyanide by C. violaceum is affected by glycine and oxygen concentrations as well as pH and temperature (Castric, 1975, Castric, 1994) with a near neutral pH showing higher cyanide production (Campbell et al., 2001). The α-carbon of glycine is responsible for the stimulation of cyanide production by cyanogenic bacteria (Brysk et al., 1969, Ward and Thorn, 1966).
It is also known that the cyanide producing bacteria work by oxidative decarboxylation as (Knowles and Bunch, 1986, Blumer and Haas, 2000). From Equation (2), it can be deduced that glycine is the direct antecedent of cyanide that is metabolised by proteobacteria such as C. violaceum. Rodgers and Knowles (1978) showed optimal production of cyanide by C. violaceum when it was grown on minimal medium that contained 10 mM glutamate, 2–3 mM glycine and 0.2–0.5 mM methionine. The Fe2+ concentration also increased the cyanide production with concentrations of 30 µM FeSO4 being used. The phosphate concentration of 68 mM was determined to be optimal for the production of cyanide by C. violaceum. However, this phosphate concentration seemed to inhibit the growth of P. aeruginosa as concentrations of>10 mM were inhibitory. In order to adapt the microorganisms to the concentrate, small amounts of concentrate (0.1 g) are generally added to the bacterial culture over a few months and the culture then transferred biweekly to sterile nutrient medium (Yopps and Baglin, 1991). The biogenic cyanide may be harvested by centrifugation of bacterial cells at 10 000 rpm for 15 min. The supernatant may be filtered and stored at 4 °C before being analysed for cyanide concentration and pH. Generally, it is not necessary to store the supernatant at 4 °C if measurements are to be done immediately after centrifugation. However, if the measurement equipment is not ready or the supernatant has to be sent somewhere else then there is need for refrigeration to prevent regrowth of cells.(2)
Liu and co-researchers (2016) observed that a mixed culture of Pseudomonas aeruginosa and C. violaceum showed the highest bioleaching capabilities. Shin and co-workers (2015) conducted a study on bio-cyanidation of PGMs from spent automotive catalysts. They produced cyanide in a continuous culture with the use of C. violaceum. The maximum biogenic cyanide concentration produced was 6594.5 mg/L (about 20 times higher than typically required for leaching free-milling gold). The authors used an air-purged C. violaceum culture bottle for biological cyanide production and two NaOH traps to capture and accumulate the HCN produced in the culture bottle according to Equation (3). The HCN is produced by the enzyme HCN-synthase that exists in Pseudomonasspecies and in C. violaceum (Blumer and Haas, 2000). The enzyme HCN-synthase produces HCN in the presence of amino acids such as glycine (Castric, 1977). The hypothetical pathway for the production of cyanide from glycine by bacteria is shown in Equation (4) (Wissing, 1974).3)HCN + NaOH → NaCN + H2O(4)
5.3.3. Potential of biogenic cyanide in PGM leaching
Based on the findings by Mwase and co-workers (2014), with chemical cyanide, bioleaching of PGMs might have to be conducted over a period of not less than 21 days. Using biogenic cyanide, Shin and co-researchers (2015) found a pH ∼ 11.8 as optimum during the leaching of Pt from secondary sources. It is known that cyanide needs to be in an environment of pH 10 or higher in order to avoid the production of HCN (Yopps and Baglin, 1991). Huang and co-researchers (2006) used a temperature of 160 °C, 10 g/l NaCN, pressure of 1.8 MPa and a solid-to-liquid ratio of 1:4 for the extraction of PGMs after removal of base metals with H2SO4, and achieved an overall recovery of 95% after 2 h. Chen and Huang (2006) also obtained the same recoveries after 1 h at the same temperature and solid-to-liquid ratio as those for Huang and co-workers (2006), but using a NaCN concentration of 6.25 g/l and pressure of 1.5 MPa. It can be deduced that Chen and Huang (2006) used less aggressive conditions than those by Huang and co-workers (2006). The effect of temperature on platinum recovery from spent industrial dehydrogenationcatalyst using cyanide leaching is shown in Fig. 6 (Shams et al., 2004). It can be deduced from Fig. 6 that highest Pt recoveries (80%) were achieved at temperatures of about 140 °C, beyond which a general decrease was observed. Higher recoveries might be achievable at lower particle sizes, higher NaCN concentrations and longer leaching during durations.