Keywords
Introduction
Direct sodium borohydride fuel cells (DBFCs) operate at close to twice the voltage of polymer electrolyte membrane fuels (PEMFCs) and yield high-power densities.1 High-voltage operation is enabled by the fact that the standard thermodynamic cell voltage of a NaBH4/H2O2 DBFC is 3.01 V, a significant enhancement over the 1.23 V theoretically obtainable with a traditional H2/O2cell (refer to Note S1 for the standard half-cell and overall reactions). The fuel, NaBH4, has a high theoretical energy density of 9.3 kWh kg−1 and specific capacity of 5.67 kAh kg−1, offering considerable advantages over competing technologies.2 Typical fuel streams include dissolved KOH to stabilize NaBH4,leading to lower practical energy densities. Dissolved H2O2 (stabilized in an acidic solution) serves as a safe and energy-dense oxidant.
A critical bottleneck in the development of these high-voltage DBFCs has been the need to stabilize NaBH4 in an alkaline solution and the H2O2 oxidant in an acidic solution to prevent the disproportionation of both species.3 Maintaining the anolyte-catholyte pH differential during operation requires an effective separator architecture. In the past, DBFCs have been primarily demonstrated with a uniform pH across the cell using either a cation exchange membrane(CEM)4, 5, 6 or an anion exchange membrane (AEM)7, 8, 9, 10 separator. The constraints introduced by the lack of a pH differential between the catholyte and anolyte have hindered these cells from achieving the potential cell voltage of 3.01 V either due to fuel and oxidant crossover or due to the use of O2 as the oxidant.
We have successfully demonstrated high-voltage NaBH4/H2O2 DBFC by maintaining a sharp pH differential using a pH gradient-enabled microscalebipolar interface (PMBI).11 The PMBI consists of a 175-μm Nafion CEM that can impede the permeability of OH− and BH4− from anode to cathode and a very thin (∼8 nm) triblock copolymer-based anion exchange ionomer (AEI) that serves as a proton barrier.11 The AEI serves as the anode binder, covers the anode catalyst, and is in intimate contact with the CEM separator, thereby forming the PMBI. The PMBI configuration facilitates maintaining a sharp local pH gradient (0.82 pH units/nm on average) at the borohydride oxidation reaction (BOR) electrocatalytic site. A state-of-the-art current density of 330 mA cm−2 at 1.5 V and a peak power density of 630 mW cm−2 at 1.0 V were obtained by incorporating this configuration in a DBFC. However, it should be noted that the system using a PMBI cannot achieve the 3.01 V theoretical cell voltage due to the unavoidable penalty from water junction potential at the interface between the AEI binder and the CEM separator.11, 12, 13
However, the system exhibited a relatively low Faradaic efficiency of 50% due to the parasitic evolution of H2 by the hydrolysis of NaBH4.11 The H2 evolution results in a mixed potential at the anode due to the occurrence of both the BOR (E0 = −1.24 V versus standard hydrogen electrode [SHE]) and the hydrogen oxidation reaction (HOR; E0 = −0.83 V versus SHE), lowering the overall cell potential. The H2 surface coverage reduces the available sites for the BOR, effectively deactivating the catalyst. Electrocatalysts exhibiting a combination of high BOR activity and selectivity (by inhibiting BH4− hydrolysis, and hence inhibiting HOR) would result in higher faradic efficiency and have been the subject of sustained interest.14, 15, 16, 17, 18, 19, 20, 21 Here, we examine an alternate reactant-transport engineering approach to improve the overall cell potential and cell performance of the DBFC.
Our reactant-transport engineering approach has examined the impact of reactant flowrate, flow velocity, residence time, and flow regime (via the Reynolds number [Re]) on DBFC performance. Microscale bipolar interfaces are coupled with different flow fields (FFs) in a DBFC to deliver practical current densities at 1.5 V. A current density of 470 mA cm−2 at 1.5 V and a peak power density of 890 mW cm−2 at 1.1 V are obtained in a PMBI-based DBFC with a 3-channel serpentine FF (3CS-FF) on the reactant side. Moreover, a PMBI-based DBFC with a 1CS-FF yields a high DBFC open circuit voltage (OCV) of 2.02 V. Balancing the competing demands of high residence time to improve BOR rates with high flowrates to detach adsorbed H2, we identify a critical Re and Damkohler number (Da) to efficiently exclude H2 gas bubbles while maintaining large current densities during the operation of a DBFC.
Results and Discussion
DBFC Historic Trends
Figure 1 summarizes the yearly progression between 2006 and 2019 in the DBFC performance state-of-the-art, using O2, air, and H2O2 oxidants.11,22, 23, 24, 25, 26, 27, 28 The key advantage of the liquid-fed DBFC over the H2/O2 gas-fed fuel cell is its high cell-operating voltage, with an ability to produce practically useful current densities at 1.5 V. However, few reports exist of DBFCs with OCVs >1.8 V since most configurations use either anion or cation exchange separators that allow facile fuel-oxidant intermixing.23,25,29 This is reflected in the black columns in Figure 1, which represent state-of-the-art (for the respective year) DBFCs that yield no current at 1.5 V. The few blue columns in Figure 1 represent reports of state-of-the-art DBFCs yielding currents at 1.5 V. Engineering the anode-separator interface to ensure an alkaline environment at the anode is imperative for DBFCs to achieve high OCV values, as the BOR exhibits a more negative potential at high pH and hence is competitive with the HOR. Thus, the measured and reported OCVs from prior studies were understood to be a mixed potential with contributions from both reactions, with a strong pH dependence.

Figure 1. Summary of the Representative Literature for DBFC Performance with O2, Air, or H2O2
The figure summarizes OCVs of the representative DBFC performance in green and current density at 1.5 V in orange. DBFCs with peak power density at high voltage (>1 V) are represented by blue columns and those with peak power density at low voltage (<1 V) are represented by black columns. The present article’s work is highlighted by the yellow column.
The column with the red hexagonal star represents our previous system incorporating the PMBI configuration in which we obtained a current density of 330 mA cm−2 at 1.5 V, a peak power density of 630 mW cm−2 at 1.0 V, and an OCV of 1.95 V.11 Building on that result, we investigated an efficient way to exclude H2 gas bubbles while maintaining a large residence time during DBFC operation. A state-of-the-art DBFC with an OCV of 1.96 V and a current density of 470 mA cm−2 at 1.5 V were obtained. A peak power density of 890 mW cm−2was achieved at 1.1 V. Using the reactant-transport engineering approach detailed below, we have exceeded our own benchmark performance by a factor of 1.4 (column highlighted in yellow in Figure 1).
Considerations and Challenges in High-Power DBFCs
Figure 2 depicts a DBFC with the PMBI configuration. The AEI covering the surface of the anode catalyst provides an alkaline environment for the BOR, with the pH value at the 40 wt% Pd/C catalyst surface being 13.5.11 During cell operation, H2O at the AEI-CEM bipolar interface dissociates into H+ and OH− to complete the overall electrical circuit. Water dissociation results in an unavoidable junction potential of −0.83 V versus SHE, resulting in a theoretical cell maximum voltage of 2.18 V, as opposed to the 3.01 V expected from only the cathodic and anodic reactions (detailed calculations provided in Note S1). A second source of OCV loss is the prevalence of side reactions and the resultant mixed potentials at the anode. To maximize the voltage efficiency of a DBFC and concurrently maintain high current densities, the anode transport regimes were engineered by using different FF architectures. The various FFs examined are depicted in the inset in Figure 2, including 1CS-FF (Figure 2A), 3CS-FF (Figure 2B), interdigitated FF (ID-FF) (Figure 2C), and pass-through FF (PT-FF) (no flow channels) (Figure 2D). Cathodic FFs that supply the reactants for the hydrogen peroxide reduction reaction (HRR) were kept unchanged, and the 3CS-FF was used in all of the cases. The same range of fuel volumetric flowrateswas used across the FFs, with the variation in the cross-sectional area of the FFs, allowing us to examine the effect of flow velocity on DBFC performance. In situstability of this DBFC configuration and ex situ stability of the anode and cathode electrodes have been evaluated and shown to satisfy operational requirements.11

Figure 2. General Cell Mechanism of the pH Gradient-Enabled Microscale Bipolar Interface-Based DBFC
The PMBI is located at the junction between the cation exchange membrane (CEM) and the anion exchange ionomer (AEI) binder. The reaction sites on the catalyst particle surface are covered by the AEI binder, which allows for selective transport of hydroxide ions. Thus, the local conditions at the reaction sites are alkaline irrespective of bulk conditions at and beyond the CEM. The flow field (FF) of the cathode is a 3-channel serpentine FF, and the FFs of the anode are varied.
(A) Single-channel serpentine FF at the anode and 3-channel serpentine FF at the cathode.
(B) Three-channel serpentine FF at the anode and 3-channel serpentine FF at the cathode.
(C) Interdigitated FF at the anode and 3-channel serpentine FF at the cathode.
(D) Pass-through (no flow channel) FF at the anode and 3-channel serpentine FF at the cathode.
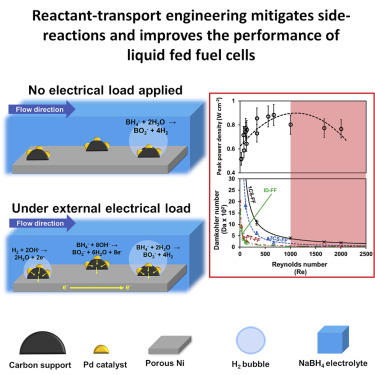
Figure 3 summarizes the challenges inherent in the BOR. For the purposes of our discussion, the anode consists of a porous Ni foam supporting a 40 wt% Pd/C catalyst (the choice of catalysts is discussed in greater depth in our earlier work11). Upon initiating fuel flow into the cell with no external electrical load (Figure 3A), the hydrolysis of borohydride is catalyzed by Pd. This results in the formation and adsorption of H2 bubbles on the catalyst surface, reducing the available catalytic sites for other reactions. Both at OCV and when an external electrical load is applied (Figure 3B), a mixed potential is generated due to the occurrence of both the BOR and HOR in parallel. Increasing the fuel flowrate is expected to drag away the adsorbed H2 bubbles and increase the availability of active sites. Nevertheless, in the absence of kinetic selection of the BOR versus HOR through a selective catalyst, competition between BH4− hydrolysis, BOR, and HOR still exists, with the trade-off between the rate of each reaction and the residence time of the fuel determining the overall cell performance. Furthermore, the present considerations of reaction competition and flow-based removal of undesired adsorbates is predicated on the existence of well-developed laminar flow and diffusion-dominated transport across a boundary layer. The sensitivity of the DBFC performance to the anolyte Reindicates that transitions in the flow regime can occur even at relatively low flowrates, and DBFC performance is not bound to increase merely with the increasing flowrate. To summarize, the key considerations when optimizing the BOR electrode are 3-fold. First, it is imperative to limit the contact duration between the catalyst and the electrolyte to limit the chemical dissociation of the NaBH4 to produce H2. Second, in direct opposition to this, it is desirable to increase the contact duration between the electrolyte and electrocatalyst to improve the reactant utilization in the DBFC. Third, the formation, nucleation, and adhesion of H2 gas bubbles on the catalyst surface limit the available sites for the desired BOR, and hence it may be desirable to increase electrolyte velocity to remove the adhering bubbles. Nevertheless, the loss of H2 will compromise the faradaic efficiency, and so the judicious design and selection of a bifunctional BOR-HOR catalyst for the anode is desirable. Thus, designing a high-performance DBFC requires the careful balancing of these considerations. This is illustrated when comparing the performance of DBFCs with different anodic FF architectures.

Figure 3. The Anode Reactions of the DBFC
(A) Electrochemical and chemical reactions at the anode of a typical DBFC without external electrical load being applied.
(B) Electrochemical and chemical reactions at the anode of a typical DBFC with external electrical load being applied. The anode consists of Pd/C catalyst supported on porous Ni foam.
Reactant-Transport Engineering Approach
Figure 4 depicts the best polarization performance of DBFC cells incorporating the various FFs under consideration on the BOR side. The cell with a 3CS-FF on the BOR side was found to exhibit the best performance in terms of the voltage and peak power density. The OCV of that cell was 1.96 V, and a peak power density of 890 mW cm−2 was obtained. A current density of 470 mA cm−2 was achieved at 1.5 V (corresponding to a power density of ∼705 mW cm−2). However, a DBFC with 1CS-FF exhibited the highest OCV, 2.02 V. The focus in the present work is on achieving and sustaining high power densities at operationally relevant voltages (i.e., >1.4 V). As seen in Figures S1–S4, increasing the fuel and reactant volumetric flux from 0.8 mL min−1 cm−2 (20 mL min−1) to 2.4 mL min−1 cm−2 (60 mL min−1) produced an increase in peak power density by mitigating mass-transport limitations at higher current densities. This linear correlation between increasing flowrates and peak power density was consistently observed for 3CS-FF, ID-FF, and PT-FF even at higher flowrates (i.e., 100 and 120 mL min−1) but was seen to break down in the case of the 1CS-FF, in which a flux of 2.4 mL min−1 cm−2 (60 mL min−1) was found to deliver the highest power. The varying cross-sectional areas of these FFs (listed in Table 1) suggested that correlating the power density to the flowrate may be more appropriate, but it was observed that while 3CS-FFs delivered their best performance at 4.8 mL min−1 cm−2 (120 mL min−1), the 1CS-FFs delivered their best performance at 2.4 mL min−1 cm−2 (60 mL min−1), despite having only one-third of the cross-sectional area. All of the comparisons were made at the same flowrates (but varying volume fluxes) so as to identify the optimum configuration for best performance at the same pump power consumption,

Figure 4. Best Polarization Curves of DBFC
The flowrate for the best performance of the DBFC with single-channel serpentine FF is 2.4 mL min−1 cm−2. The flowrate for the best performance of the DBFC with 3-channel serpentine FF is 4.8 mL min−1 cm−2. The flowrate for the best performance of the DBFC with interdigitated FF is 4.8 mL min−1 cm−2. The flowrate for the best performance of the DBFC with pass-through FF is 4.0 mL min−1 cm−2.
Table 1. Reactant-Transport Characteristics of FFs Used in DBFC
| Type of FF (volumetric flow rate, mL/min | Flow velocity, m/s | Re | Residence time through flow channel, s | Da × 102 | Mean resident time through electrode, s | |
|---|---|---|---|---|---|---|
| single-channel serpentine (areaC/S = 1 mm2) | ||||||
| 20 | 0.33 | 334 | 4.83 | 10.73 | 170 | |
| 60 | 1 | 1,002 | 1.61 | 3.93 | 56.67 | |
| 100 | 1.67 | 1,670 | 0.97 | 2.28 | 34 | |
| 120 | 2 | 2,004 | 0.81 | 1.88 | 28.33 | |
| 3-channel serpentine (areaC/S = 3 mm2) | ||||||
| 20 | 0.11 | 111 | 4.83 | 18.46 | 275 | |
| 60 | 0.33 | 334 | 1.61 | 6.17 | 91.67 | |
| 100 | 0.56 | 557 | 0.97 | 3.77 | 55 | |
| 120 | 0.67 | 668 | 0.81 | 1.96 | 28.33 | |
| interdigitated (areaC/S = 15 mm2) | ||||||
| 20 | 0.022 | 22 | 2.16 | 7.81 | 175 | |
| 60 | 0.067 | 67 | 0.72 | 3.61 | 58.33 | |
| 100 | 0.11 | 111 | 0.43 | 2.25 | 35 | |
| 120 | 0.13 | 134 | 0.36 | 1.86 | 28.33 | |
| pass through (areaC/S = 50 mm2) | ||||||
| 20 | 0.007 | 25 | 7.5 | 9.00 | 250 | |
| 60 | 0.020 | 76 | 2.5 | 4.32 | 83.33 | |
| 100 | 0.033 | 127 | 1.5 | 2.53 | 50 | |
Da, Damkohler number; FF, flow field; Re, Reynolds number.
Figure S5A depicts the effect of the electrolyte velocity on the OCV. The measured OCV is a mixed potential with contributions from the BOR and HOR. The promise of high operational voltages in a DBFC can be achieved only if the BOR is the dominant reaction. The chemical decomposition of NaBH4 to produce H2 that is subsequently oxidized to water renders the system a H2/O2 or H2/H2O2 fuel cell, with NaBH4 being the H2 storage media (the reason for the ≤1 V OCVs observed in several purported DBFCs from the literature). The selectivity30 of the BOR catalyst used in the present system means that the major factor lowering the OCV is the blocking of BOR reaction sites by adsorbed H2 bubbles. Thus, at the low velocity limit, we see dramatic gains in the system OCV, as small gains in flow velocity significantly aid in H2 bubble removal.31, 32, 33 Given the low velocity needed to remove these bubbles, this effect is not as pronounced at high velocities, and no gains in the OCV are seen with increasing fluid velocity >0.5 m s−1.
Figure S5B depicts the effect of the flow velocity on the peak power density. The peak power density is influenced by the BOR selectivity of the catalyst. The peak power density increases with increasing velocity as the faster flowing fluid removes H2 bubbles before they have a chance to undergo HOR and additionally frees up reaction sites for the BOR. Above 1 m s−1 in cases of the 1CS-FF, the peak power was observed to decrease. This is due to the transition of the flow regime from laminar to turbulent, Re approaching 2,000. The plateauing of the peak power value over 0.4 m s−1 indicates that this value of flow velocity is sufficient for bubble detachment, while the decrease after 1 m s−1 indicates that the flow regime significantly affects the BOR.
Figure S5C depicts the effect of the channel residence time (τchannel) on the peak power density. Higher τchannel will result in greater chemical production of H2(limited only by the availability of adsorption sites), which will in turn compete with the BOR. Thus, we see that in the case of the 3CS-FF, PT-FF, and ID-FF, the peak power density is inversely corelated with the residence time. While this inverse trend is observed in the case of the 1CS-FF for high residence times (low fluid velocity), the correlation breaks down at high fluid velocities (low residence times) due to the transition in the flow regime. Despite the 3CS-FF and 1CS-FF exhibiting the same residence times, the observed divergence in cell performance indicates that τchannel is not the primary factor affecting the performance of DBFC.
Following the observation that the electrolyte flow velocity (and its corollary, the residence time) cannot completely account for the observed DBFC performance, we turned to the dimensionless Da, which is the ratio of the reaction rate to the transport rate. Given the variety of possible rate law expressions, Da34 can be defined in general as:(Equation 1)where k is the rate constant for a nth order reaction with units of (mol cm−3)(1−n)s, is the initial (inlet) concentration of the reactant (1.5 M NaBH4 in 3 M KOH), τ is the porous anode (Ni foam) residence time in seconds, and r is the rate of an nth order reaction in mol s−1. Given that i = nFAr and PPeak = ipeakEpeak, where PPeak is the peak power, iPeak is the peak current, and EPeak is the potential corresponding to peak power, we obtain the following expression for the Da for a DBFC:(Equation 2)Da captures the effect of the peak power, the residence time through the electrode, and the reactant concentration in one term. An example calculation of Da is provided in Note S1. The residence time in the anode catalyst layer was measured by a residence time distribution (RTD) measurement using a step tracer input (representative inlet and exit tracer concentrations provided in Figure S9), as detailed in the Experimental Procedures. Figure 5 depicts the plots of (1−F) versus t. , where t at (1−F) = 0 is taken to be the anode catalyst layer residence time (τ) and used in the calculation of Da. The effect of the FFs on the residence time in the catalyst layer was examined by comparing τchannel with τ. τchannel was calculated by dividing the volume of the flow channels by the volumetric flowrate. τchannel and τ were found to be linearly correlated, as depicted in Figure S10. Thus, the FF channel volume will allow us to directly control the residence time in the catalyst layer.

Figure 5. Reactant Residence Times at DBFC Anodes
(A–D) Cumulative distribution curves of DBFC with (A) single-channel serpentine FF at the anode and 3-channel serpentine FF at the cathode, (B) 3-channel serpentine FF at the anode and 3-channel serpentine FF at the cathode, (C) interdigitated FF at the anode and 3-channel serpentine FF at the cathode, and (D) pass-through (no flow channel) FF at the anode and 3-channel serpentine FF at the cathode.
Given that the effect of the flow velocity is dependent on the architecture of the flow path, comparison between FFs is best carried out using an architecture agnostic criterion such as Re. Re35 is defined as:(Equation 3)where ρ is the density of the fluid in kg m−3, u is the fluid velocity in m s−1, L is the characteristic length in m, and μ is the dynamic viscosity in Pa s−1. The characteristic length (hydraulic diameter) for a square duct (in the case of the serpentine and interdigitated FFs) is the side of the square, while it is given by the following equation in the case of the rectangular cross-section35 for the pass-through FF:(Equation 4)where a is the width of the rectangular cross-section and b is the length of the rectangular cross-section. Da was found to be related to the Re by an inverse power law, as depicted in Figure 6. The peak power was found to decrease with increasing Re beyond 1,000, as seen in Figure 6. The increasing Re indicates faster reactant transport and hence should lead to a decrease in Da. Given the difference in the characteristic times for reaction and transport, the non-linear decrease in Da is along expected lines.34 However, increasing Re was also found to lead to a decrease in the peak power in the region of transition between laminar and turbulent flow regimes. To explain this observation, we examined the influence of the Re on the rate of the BOR.

Figure 6. Reactant-Transport Engineering at the DBFC Anode
Effect of the Reynolds number on the Damkohler number and the peak power density.
The error bars represent the standard error, with n = 3.
Full Cell Impact of Anode Reactant-Transport Engineering
The OCV and the peak power density are characteristics of the full cell, while the effect of the changes in the FF is being examined on the anode. Assuming that the reaction at the cathode is only the reduction of H2O2, the half-cell voltage at the anode (Eanode) can be calculated from the OCV after accounting for the bipolar junction potential. The relationship11 is as follows:(Equation 5)Here, E0HRR = 1.77 V versus SHE and Ej = 0.83 V versus SHE. The anodic mixed potential consists of contributions from BOR and HOR (with the H2 being chemically produced by the hydrolysis of the NaBH4):(Equation 6)The BOR efficiency (ηBOR) versus Re is depicted in Figure S6. To deconvolute the effect of the reactant flow on the anodic reactions, we elucidated the contributions from the BOR and the HOR to the cell current.
Figure 7 depicts the variation of the BOR rate with Re. Given that the overall cell current density (here, the current density at peak power is considered) has contributions from the BOR and HOR, we obtain the following relationship:(Equation 7)which in turn may be written as:(Equation 8)Given nBOR = 8, nHOR = 2, F (Faraday’s constant) = 96,485 C mol−1, and rBOR/(rBOR + rHOR) = ηBOR, the rBOR depicted in Figure 7 was calculated. The rBOR was found to increase with increasing Re as the higher velocities aid in the removal of adsorbed H2 bubbles and increase the number of surface sites available for BOR. This effect was found to plateau over Re = 1,000, indicating that the reaction is not limited by the availability of NaBH4. Upon further increasing the flow velocity, the peak power was found to decrease. At these high flowrates, τchanneland τ have similarly small values and may indicate poor penetration of the reactant flow into the catalyst layer, accounting for the decrease in peak power. The correlation between Re and rHOR (Figure S7) was found to be weak, as the H2bubbles are already present at the surface and hence do not need higher flow velocities to clear out surface sites. H2 production by the chemical decomposition of NaBH4 is dependent on the contact time with the catalyst. In summary, we recommend that high-power DBFCs be designed to keep the Re of the flow within their anodic channels of between 300and 1,000 and ensure a Dabetween 2 × 10−2 and 5 × 10−2.

Figure 7. Impact of the FF Regime
Relation between Reynolds number and borohydride oxidation reaction (BOR) rate and polynomial fit of experimental data from all FFs.
The error bars represent the standard error, with n = 3
Extending and Supplementing Reactant-Transport Engineering
At the stack level, we caution that one cannot indefinitely increase the flow velocity without eventually compromising the overall system efficiency. A clear analog is seen in the case of PEMFCs. Chen et al. built a PEMFC system model incorporating the stack, heat exchanger, water tank, cooling pumps, and gas-processing components. Upon increasing the hydrogen (anode reactant) pressure from 1 to 3 atm, the electric power output rose from 3.5 to 7.7 kW, while the system efficiency only increased from 73.6% to 76.2%. Thus, Chen et al.concluded that the power output of a fuel cell stack is not correlated with the system efficiency.36 In a DBFC, the flow velocity of NaBH4 cannot be increased indefinitely due to both the observed decrease in peak power at higher Re and the possible decrease in overall system efficiency when accounting for pumping power requirements.
The faradaic (fuel) efficiency of BOR is usually determined by electrochemical or spectro-electrochemical methods in model conditions (well-defined noble electrocatalyst and dilute anolytes). The use of dilute BH4− solutions for fundamental studies mitigates interference from gas bubble formation and electrode passivation by boron-oxide precipitation. However, the faradaic efficiency measured in these model experiments (in dilute NaBH4 solutions) are not readily applicable under DBFC operating conditions (in concentrated NaBH4solutions). Studies with higher concentrations are precluded due to the occurrence of unwanted side reactions and challenges in maintaining a clean electrode surface due to bubble adhesion and side-product passivation.21 The development of selective catalysts that promote the BOR and HRR while inhibiting the hydrogen and oxygen evolution reactions (and hence improving faradaic efficiency) and systematic analysis of overall cell efficiency are valuable next steps in the development of DBFCs.
The present discussion about Da number, Re number, and BOR rate is also applicable to DBFCs operating with other oxidants or without the PMBI. However, in DBFCs operated with AEM or CEM separators, the crossover of BH4from anode to cathode or of H2O2 from cathode to anode, respectively, results in the loss of fuel and oxidant, and a lowering of the operating voltage thereby adversely affects performance.30 Thus, attempts to improve the performance of those DBFCs using the transport engineering approach may only yield limited gains in performance due to the potentially far more significant effect of the fuel-oxidant crossover.
Comparison with a PEMFC
The state-of-the-art DBFC performance using the PMBI configuration with 3CS-FF is compared with a PEMFC (H2/air, unpressurized)37 in Figure 8. The PEMFC yielded a power density of ∼295 mW cm−2 at 0.75 V, whereas our DBFC with the PMBI configuration provides a 2.4 times higher power density (705 mW cm−2) at double the operating voltage (1.5 V). At the same operating current density (500 mA cm−2), the voltage efficiency of our DBFC is ∼78%, whereas the voltage efficiency of a PEMFC is ∼57%. The high-voltage operation in conjunction with high power density confers twin advantages—it reduces the cost of fuel cell stacks by downsizing and considerably simplifying the design, and doubling the cell voltage potentially halves the number of cells. This is achieved without affecting power output, as current is proportional to cell area, which can be readily scaled. Thus, DBFCs can be seamlessly integrated or retrofitted into propulsion systems for weight-sensitive transportation platforms such as submersibles and drones.