1. Introduction
Self-assembly has long been used as a standard approach for the synthesis of functional matter. [1], [2], [3], [4] Among all, crystals are a particularly important class of self-assembled materials; they represent the best case scenario to induce large-range ordered structures with control at the molecular level. [5,6] To date, this control is accomplished following a rational design of the molecular building units, where supramolecular synthons (such as non-covalent bonds) are being used to ensure specific recognition and interaction events that ultimately lead to a desired thermodynamic crystalline product (i.e. equilibrium state). Thermodynamic products reside in the global minima of Gibbs energy, [7] a state where it becomes almost impossible to tune crystals’ function. In this context, controlled defect engineering has emerged as an advance tool to modify crystals’ properties. [8] Defects have shown to have a decisive impact on changing the physicochemical properties of functional thermodynamic crystalline products, e.g. novel sorption and catalytic activities have been reported. [9]
Therefore, in crystal engineering research, much attention is being focused on either controlling the spatial distribution of defects in crystals [10] or on harnessing their aggregation pathway (controlling pathway complexity). [11]Note that the latter can ultimately achieve the isolation of non-equilibrium crystal states (e.g. kinetically trapped or metastable species) that can display different structures and functions. The most prevalent method to create defects in crystals is the incorporation of building block fragments in the reaction mixture that locally break the regularity of the thermodynamic crystal structure. [8] On the other hand, crystallization protocols involving short reaction times are frequently used to control crystallization kinetics. [12], [13], [14] Note that, in spite of their success, both approaches (i.e. the controlled synthesis of defects and non-equilibrium states) are still challenging and yet are essential for future advances in chemistry, materials science as well as nanotechnology. Notably, achieving defects in crystals through non-equilibrium crystal states is so far unexplored, but it is a key step to unveil novel structure-property correlations that can lead to unprecedented properties and applications.
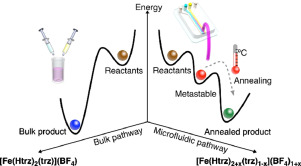
Herein, we demonstrate that pathway selection to an unprecedented non-equilibrium crystal state (i.e. a metastable state, Fig. 1e) can be successfully accomplished when the interplay of a chemical reaction with the mass transport of reagents can be harnessed in space and time. Specifically, we prove that controlled reaction-diffusion (RD) conditions achieved inside a continuous flow microfluidic device [15] can ultimately allow a kinetic control synthesis of a functional coordination polymer (CP) incorporating charge imbalance defects in its structure (i.e. protonation defects). Further, we show that the annealing of this non-equilibrium crystal state (i.e. the metastable state) yields a new thermodynamic product (Fig. 1e, compare bulk pathway with microfluidic pathway). We focused on the well-known iron-triazole complex [Fe(Htrz)2(trz)]n(BF4)n (Htrz = 1,2,4−triazole; trz− = 1,2,4−triazolato), which exhibits spin-crossover behavior (hereafter SCO-1). [16,17] We show that while the bulk synthesis of SCO-1 consistently yields crystalline microparticles(hereafter bSCO-1) of the extensively reported thermodynamic product, [18] the synthesis under controlled RD conditions leads to sub-30 nm crystalline particles of a non-stoichiometric form of SCO-1 (hereafter RD-SCO-1) characterized by a non-stoichiometric triazole/triazolato ratio. Remarkably, the spin transition behavior of RD-SCO-1 is radically different to the one observed for bSCO-1. The latter clearly shows that protonation defects can endow the formation of a new crystal structure with unique properties. Note that we chose SCO-1 for the current investigation, i.e. one of the most widely studied SCO compounds, [19], [20], [21], [22] since the switchable electronic behavior of iron(II)-triazole complexes when exposed to different external stimuli is highly sensitive to structural modifications of the crystalline network. [23] For example, the magnetic properties of this family of SCO compounds have been tuned by using different triazole derivatives, either as pure ligands or in mixtures, or different counter anions. [24] Thus, using SCO-1 represents an excellent scenario to benchmark the impact of structural defects engineered by the use of controlled RD conditions.
 Fig. 1. Schematic representations of the (a) microfluidic and (b) bulk synthesis of SCO-1. (c) Thermal behavior of the magnetic susceptibility for bSCO-1 (blue curve) and the as prepared RD-SCO-1 (red curve). The magnetic susceptibility curve of the annealed RD-SCO-1 is presented in green. (d) Photograph of RD-SCO-1 powder re-dispersed in water showing the color change upon the LS → HS spin transition. (e) Schematic illustration of the bulk and microfluidic reaction pathways yielding to stoichiometric bSCO-1 and non-stoichiometric RD-SCO-1, respectively. Along the microfluidic pathway a metastable state is first formed, which then relaxes to a thermodynamic equilibrium state upon thermal annealing. (f) Molecular structure of the 1D coordination chain of SCO-1.
Fig. 1. Schematic representations of the (a) microfluidic and (b) bulk synthesis of SCO-1. (c) Thermal behavior of the magnetic susceptibility for bSCO-1 (blue curve) and the as prepared RD-SCO-1 (red curve). The magnetic susceptibility curve of the annealed RD-SCO-1 is presented in green. (d) Photograph of RD-SCO-1 powder re-dispersed in water showing the color change upon the LS → HS spin transition. (e) Schematic illustration of the bulk and microfluidic reaction pathways yielding to stoichiometric bSCO-1 and non-stoichiometric RD-SCO-1, respectively. Along the microfluidic pathway a metastable state is first formed, which then relaxes to a thermodynamic equilibrium state upon thermal annealing. (f) Molecular structure of the 1D coordination chain of SCO-1.2. Results and discussion
Fig. 1a shows a schematic of the four-inlet microfluidic device used for the synthesis of RD-SCO-1. In a typical experiment, two ethanol solutions of Fe(BF4)2•6H2O (1 M) and Htrz (3 M) were injected respectively through the middle channels at a flow rate of 50 µL/min, while pure ethanol was injected in the two side channels at a flow rate of 100 µL/min. That is, a flow rate ratio (FRR) of 2 was established during the microfluidic synthesis. Hereafter, we define FRR as the ratio between the ethanol sheath flows and the reagent-laden flows. The reaction mixture coming out from the outlet, i.e. once the microfluidic reaction was completed, was quenched by dilution in pure ethanol and immediately centrifuged to avoid the continuing of the reaction out-of-chip. This procedure afforded milligram quantities of a purple powder, i.e. of RD-SCO-1 complex (Figure S1 and Experimental Section). As a comparison, bSCO-1 was prepared by a standard bulk preparation method from the same two ethanolic solutions of precursors (Fe(BF4)2•6H2O and Htrz) used for the microfluidic synthesis (Fig. 1b and Experimental Section).
Variable-temperature magnetic susceptibility measurements for complexes RD-SCO-1 and bSCO-1 revealed a radically different spin transition behavior. Fig. 1c shows the χT vs temperature (T) curves (where χ is the molar paramagnetic susceptibility per Fe(II) center) of the two complexes. While bSCO-1 exhibits a strongly cooperative spin transition at 384 K (Tc↑) from the low spin (LS) to the high spin (HS) state, accordingly with that reported in literature [18] for this material with a large hysteresis upon cooling (Tc↓ = 344 K, ΔT = 40 K), RD-SCO-1 features a much less cooperative LS → HS transition, with a more gradual change of χT vs T at lower temperature and an abrupt change only around 369 K. In addition, and unexpectedly, the hysteresis width observed in the first thermal cycle (ΔT = 46 K) is dramatically reduced in the second (ΔT = 25 K) and third thermal cycles (ΔT = 18 K) for RD-SCO-1, while it remains practically unaltered for bSCO-1 (Fig. 1c shows the first and third cycles and Figure S2). Note that the narrowing of the hysteresis for RD-SCO-1 is primarily due to a reduction of Tc↑ in the LS → HS transition (warming branch) with successive thermal cycles. Table 1 summarizes the transition temperatures for RD-SCO-1 and bSCO-1 complexes.
Table 1. Transition temperatures of SCO-1 complexes obtained from magnetic measurements for consecutive cycles.
| Magnetic Measurements | ||||
|---|---|---|---|---|
| Cycle | Tc↑ (K) | Tc↓ (K) | ΔT (K) | |
| bSCO-1 | 1 | 384 | 344 | 40 |
| RD-SCO-1 (FRR2) | 1 | 369 | 323 | 46 |
| 2 | 345 | 320 | 25 | |
| 3 | 338 | 320 | 18 | |
The thermal induced spin transition from LS to HS is also accompanied by strong thermochromism; for example, RD-SCO-1 nanoparticles dispersed in water change from purple (LS) to white (HS) upon heating (Fig. 1d). In addition, variable-temperature UV–vis spectra show a decrease of intensity of a band centered at 590 nm (corresponding to the 1A1 → 1T1 transition of Fe(II) in the LS state), with the concomitant appearance of a broad band in the near-infrared region (ascribed to the 5T2 → 5E transition of Fe(II) in the HS state) upon heating above Tc↑. [25], [26], [27] Upon subsequent cooling, the spectra switch back to the original shape with evident thermal hysteresis (Figure S3), while the RD-SCO-1 nanoparticles dispersed in water turn back to purple (Fig. 1d). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of RD-SCO-1 clearly show that the microfluidic synthesis yields nanoparticles with diameters of 26 ± 9 nm (Fig. 2b-d, Figure S4 and Table S1). Note that RD-SCO-1 was deposited on TEM grids either by collecting the reaction mixture directly eluting from the chip, or by re-dispersing in ethanol the isolated powder (i.e. after quenching and centrifugation), leading to comparable results (compare Fig. 2c and Figure S4d). The latter indicating that the post-synthetic treatment does not affect noticeably the size and shape of the formed nanoparticles. In addition, DLS measurements on RD-SCO-1 nanoparticles re-dispersed in ethanol or water confirmed a narrow size distribution centered around 30 nm (polydispersity index, PDI ≤ 0.21), which is in good agreement with the dimensions extracted by SEM and TEM analysis (Fig S5 and Table S1). By contrast, the bulk synthesis of bSCO-1 results in larger plate-like crystals with sizes around 200 nm (Fig. 2a).
 Fig. 2. SEM images of (a) bSCO-1 and (b) RD-SCO-1. (c) TEM image of RD-SCO-1 powder re-dispersed in ethanol. (d) Size distribution of RD-SCO-1 nanoparticles from SEM and TEM analysis. (e) Complete XPS spectrum recorded at 298 K for RD-SCO-1. The peaks corresponding to Si2p and O1s are due to the use of siliconsubstrates for the deposition of the samples. (f) Corresponding high-resolution XPS spectrum for Fe2p with curve-fitting. A1 and B1 components correspond to Fe2p3/2 and Fe2p1/2 for the LS state, respectively; A2 and B2 are their HS satellites.
Fig. 2. SEM images of (a) bSCO-1 and (b) RD-SCO-1. (c) TEM image of RD-SCO-1 powder re-dispersed in ethanol. (d) Size distribution of RD-SCO-1 nanoparticles from SEM and TEM analysis. (e) Complete XPS spectrum recorded at 298 K for RD-SCO-1. The peaks corresponding to Si2p and O1s are due to the use of siliconsubstrates for the deposition of the samples. (f) Corresponding high-resolution XPS spectrum for Fe2p with curve-fitting. A1 and B1 components correspond to Fe2p3/2 and Fe2p1/2 for the LS state, respectively; A2 and B2 are their HS satellites.It should be noted that, to the best of our knowledge, nanoparticles of the iron-triazole complex SCO-1 have only been prepared by batch processes, such as using reverse-micelle approaches (microemulsions), which involve reaction times of several hours and the use of surfactants. [28] Nonetheless, the reverse-micelle method leads to nanoparticles of SCO-1 that exhibit comparable magnetic behavior as the bulk material, with a cooperative abrupt LS → HS transition and unmodified thermal hysteresis, even for sizes as small as 10 nm. [18,24] Conversely, by using our approach, RD-SCO-1 nanoparticles were generated within milliseconds and without the need for surfactants during the synthesis; an approach that yields an unprecedented SCO-1 complex that displays a completely different spin transition behavior. We anticipate that the differences in the magnetic susceptibility versus temperature curves observed for RD-SCO-1 and bSCO-1 cannot be merely ascribed to the different size/morphology of the respective crystals (vide infra), which is in agreement with the scarce effect of downsizing SCO-1 crystals on its magnetic propertiesextensively reported in the literature. [27,29] Thus, to shed light into the causes underlying the peculiar magnetic behavior of RD-SCO-1, we fully characterized the nanoparticulate material obtained from the microfluidic synthesis by a range of different techniques. Elemental energy-dispersive X-ray (EDX) analysis confirmed the presence of C (Kα = 0.28 keV), N (Kα = 0.39 keV) and Fe (Kα = 6.39 keV). Additionally, the presence of BF4− counterions was validated by the detection of a peak corresponding to F (Kα = 0.68 keV), thus proving the formation of the iron-triazole complex (Figure S6). X-ray photoelectron spectroscopy (XPS) was used to confirm the chemical bonding in RD-SCO-1 samples, and thus, the formation of 1D coordination chains. In Fig. 2e, the full XPS spectrum shows the C1s and N1s peaks of the imidazole ligands together with the peak corresponding to Fe(II) 2p. In addition, the presence of BF4− was confirmed by the appearance of the corresponding peaks for B and F at 195.2 eV and 686 eV, respectively. To gain more information about the Fe(II) bonding environment, high resolution XPS was performed (Fig. 2f and Figure S7). Notably, these measurements confirmed that the Fe(II) nodes retain their oxidation state during the microfluidic synthesis. As shown in Fig. 2f, the peaks corresponding to Fe2p3/2 and Fe2p1/2 along with their characteristic satellites were present in RD-SCO-1, which is a typical fingerprint of iron-triazole complexes with an octahedral coordination and a spin switching behavior. [30]For comparison, the XPS data for bSCO-1 are reported in Figure S7.
The chemical composition of both RD-SCO-1 and bSCO-1 was experimentally determined using a combination of inductively coupled plasma mass spectrometry (ICP-MS) and elemental analysis. The results are displayed in Table 2. The data obtained for bSCO-1 confirm, within the experimental error, the formation of the well-known polymeric complex with formula [Fe(Htrz)2(trz)]n(BF4)n (Fig. 1f), i.e. corresponding to the expected stoichiometry triazole/triazolate 2:1 (found: [Fe(Htrz)2.0(trz)1.0](BF4)1.0). On the other hand, and in sharp contrast, the data for RD-SCO-1 clearly indicate the formation of a non-stoichiometric form of the iron-triazole complex with a calculated ratio triazole/triazolate 2.2:0.8 (Table 2). That is, there is an excess of neutral (non-deporotonated) triazole ligand which is compensated by extra BF4−counterions, on average one out of every five iron centres along the polymeric chain. We point out that a SCO complex with formula [Fe(Htrz)3](BF4)2, i.e. containing only neutral triazole ligands has been previously prepared from the same precursors (Fe(BF4)2•6H2O and Htrz) by using different bulk reaction conditions (e.g. modulating solvent composition and reactants concentrations). [16,31] Importantly, note that here RD-SCO-1 and bSCO-1 are prepared under the same reaction conditions, the main difference is that in the former a control mass transport during the coordination reaction is established. [15] To the best of our knowledge, non-stoichiometric iron-triazole complexes comprising protonation defects are not reported in the literature. This is because, with common bulk synthetic approaches, it is a challenge to harness the ratio of triazole/triazolato species incorporated in the structure. [32] In fact, to achieve controlled protonation states in complexation reactions has proved to be a challenging problem because the proton uptake/release upon the interaction event is frequently occurring at pHs different from the optimum pH of complexation. [33] Actually, for example, under thermodynamic control only the favored stoichiometry triazole/triazolate 2:1 is obtained (Fig. 1e, bulk pathway), [24]and the engineering of protonation defects cannot thus be achieved. Therefore, we believe that the presence of such protonation defects is at the basis of the different magnetic properties displayed by RD-SCO-1 when compared to bSCO-1. In fact, the spin crossover phenomenon and degree of cooperativity of the spin transition is strongly dependent on the crystalline structure and intermolecular non-covalent interactions between the 1D coordination chains, which in turn are bound to be strongly affected by the presence of additional BF4− between them. Importantly, the chemical analysis ruled out the presence of water molecules in RD-SCO-1. Thus the narrowing of the hysteresis for RD-SCO-1 with reduction of Tc↑ after the first thermal cycle cannot be ascribed to simple solvent release upon warming, as it has been reported for other iron-triazole complexes containing water of crystallization. [31] The absence of water in RD-SCO-1 nanoparticles was also confirmed by the lack of a broad absorption band around 3600 cm−1 in the corresponding FT-IR spectrum (Figure S8). On the other hand, the FT-IR spectrum displays bands at 1536 cm−1 and 1497 cm−1 corresponding to stretching vibrational modes of protonated and deprotonated triazole ligands respectively (Figure S8). Upon heating, the main IR peaks only experience minor energy shifts and intensity variations, with the spectra reverting to the original shape once the system is completely cooled back to the LS state, thus corroborating the chemical stability of RD-SCO-1 (Figure S8). Finally, the lack of water was also confirmed by thermogravimetric (TGA) analysis showing no significant weight loss upon heating until decomposition occurred at T > 450 K (Figure S9).
Table 2. Chemical compositional formula determined by combination of elemental analysis (for C,N and H) and ICP-MS (for metal quantification) for SCO-1complexes.
| Elemental Analysis (calculated*) | Error | Chemical Formula | Percentage | ||||
|---|---|---|---|---|---|---|---|
| %C | %H | %N | %metal⁎⁎ | ||||
| bSCO-1 | 20.80 (20.66) | 2.38 (2.31) | 36.03 (36.14) | 16.00 (16.01) | 0.19% | Fe1 Htrz2 Trz1(BF4−)1 | 40% Htrz, 20% Trz, 16% Fe |
| RD-SCO-1 (FRR2) | 19.06 (19.87) | 2.56 (2.27) | 35.21 (34.76) | 15.67 (15.40) | 0.97% | Fe1 Htrz2.2Trz0.8(BF4−)1.2 | 41% Htrz, 15% Trz, 15% Fe |
- ⁎
-
The percentages in parenthesis are calculated values following the theoretical percentages and considering two constraints: complete coordination sphere and neutral charge.
- ⁎⁎
-
The values were quantified from ICP-MS.
To sum up, the results from chemical analysis, FT-IR and TGA indicate that the narrowing of the hysteresis loop for RD-SCO-1 nanoparticles with successive thermal cycles (Figure S2) can be attributed neither to desolvation [31] nor to chemical decomposition. Instead, it has to be ascribed to an annealing process of the supramolecular structure of RD-SCO-1. In other words, the synthesis under controlled RD conditions (microfluidic pathway) yields a metastable non-equilibrium state of a non-stoichiometric SCO-1 complex (i.e. a metastable state of RD-SCO-1) (Fig. 1e) characterized by a less abrupt spin transition than bSCO-1 (red and blue curves in Fig. 1c, respectively). Upon thermal annealing, the metastable state of RD-SCO-1 equilibrates to a thermodynamic one (thermodynamic RD-SCO-1 state) that features a much narrower hysteresis and a significantly lower Tc↑ (green curve in Fig. 1c).
To get more insights into this annealing process, we performed small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) experiments, as well as calorimetric measurements on the nanoparticulate powder of RD-SCO-1. Fig. 3a shows the SAXS profiles at room temperature of the as prepared RD-SCO-1, i.e. the metastable state of RD-SCO-1, and of the annealed product obtained after two heating/cooling cycles (i.e. LS →HS →LS loops). A significant decrease of the absolute scattering intensity and change of the SAXS profile at low values of the scattering vector (q < 1) are observed upon annealing. The SAXS data can be fitted to a polydisperse (Schultz-distribution) [34] compact sphere model giving an average diameter of 23.4 ± 2 nm (with polydispersity index PI = 0.44) for the metastable state of RD-SCO-1 and of 66 ± 5 nm (with PI = 0.2) for the annealed one, indicating that the annealing promotes a growth/coalescence of RD-SCO-1 nanoparticles (Figure S10). Both the increase in size and global intensity reduction in the SAXS profiles are in agreement with the reduction of particle surface. Moreover, the sizes measured by SEM for the metastable state of RD-SCO-1 (Fig. 2d) and the thermodynamic RD-SCO-1 complex (Fig. 3b) are consistent with the obtained SAXS data. While we cannot exclude, a priori, a small influence of the increase of particle size to the change of the magnetic behavior upon annealing, we believe that this is not the main contribution (see below).
 Fig. 3. (a) SAXS profiles at room temperature (LS state) for the as prepared (metastable, red) and annealed (after two thermal cycles, green) RD-SCO-1. The solid lines represent the fit to a polydisperse rigid sphere model. (b) Size distribution histogram of RD-SCO-1 after annealing. Inset shows a SEM image of annealed RD-SCO-1. (c) WAXS profiles of the metastable and annealed forms of RD-SCO-1 in the LS state (red and green, respectively) and of RD-SCO-1 in the HS state (black). (d) WAXS profiles of initial and cooled bSCO-1 in the LS state (blue and magenta, respectively) and of bSCO-1 in the HS state (black).
Fig. 3. (a) SAXS profiles at room temperature (LS state) for the as prepared (metastable, red) and annealed (after two thermal cycles, green) RD-SCO-1. The solid lines represent the fit to a polydisperse rigid sphere model. (b) Size distribution histogram of RD-SCO-1 after annealing. Inset shows a SEM image of annealed RD-SCO-1. (c) WAXS profiles of the metastable and annealed forms of RD-SCO-1 in the LS state (red and green, respectively) and of RD-SCO-1 in the HS state (black). (d) WAXS profiles of initial and cooled bSCO-1 in the LS state (blue and magenta, respectively) and of bSCO-1 in the HS state (black).The WAXS spectrum at room temperature (i.e. in the LS state) of the as prepared RD-SCO-1 powder displays weak and broad scattering peaks at 24.5° an 26.2° as expected for a crystalline material with nanometer-sized domains (Fig. 3c). [35]When the RD-SCO-1 powder is heated up to the HS state, a significant change of the WAXS profile is observed with the appearance of sharper and well-defined new peaks in the region between 22° and 25° and substantial disappearance of the broad feature at 24.5° More importantly, after annealing (two LS → HS → LS loops), the WAXS spectrum in the LS state of the thermodynamic RD-SCO-1 complex is strikingly different from that of the metastable form, displaying sharp features at 22.9°, 24°, 25°and 26.6° This behavior clearly indicates that RD-SCO-1 undergoes a structural rearrangement upon annealing. In marked contrast, the WAXS spectrum in the LS state of as prepared bSCO-1 is perfectly overlapped to that recorded at room temperature after performing LS → HS → LS thermal cycles (Fig. 3d), thus indicating that the structural changes upon spin transition are completely reversible in the case of bSCO-1. We believe that the differences in the crystalline structure observed for the metastable and thermodynamic forms of RD-SCO-1 arise from the non-stoichiometric nature of this compound and likely imply a rearrangement-redistribution of the charge defect along the polymeric chain associated with a rearrangement of the BF4−counter anions. The latter results in the modification of the hysteresis loop observed upon annealing.
Differential scanning calorimetry (DSC) measurements on RD-SCO-1 and bSCO-1 showed an endothermic peak upon heating and an exothermic one upon cooling corresponding to the LS →HS and HS →LS spin transitions respectively (Figure S11). The transition temperatures extracted from DSC peaks are in good agreement with the magnetic measurements, with only small deviations due to the different temperature scanning rates used in both techniques (Table S2). Interestingly, the calorimetric peaks for RD-SCO-1 are broader than those for bSCO-1, which is in line with the less cooperative nature of its spin transitions. Finally, Table S2 summarizes the enthalpy and entropy variation (ΔH and ΔS) estimated from the DSC curves in the different heating cooling cycles. Overall, the enthalpy variations associated with all spin transitions are lower for RD-SCO-1 than for bSCO-1. In addition, the data show that the ΔH associated to the first heating run of RD-SCO-1 (14.6 kJ/mol) is greater than that of the second one (11.8 kJ/mol). This suggests that the first heating run does not only involve the LS → HS spin transition but possibly includes also changes occurring in the supramolecular structure of the complex, which is consistent with both the WAXS and magnetic data. In sharp contrast, the enthalpy and entropy differences between different heating runs for the bulk sample are negligible (Table S2).
Finally, we explored the possibility of controlling the stoichiometry of RD-SCO-1, and thus its spin transition behavior, by modulating the FRR in the microfluidic synthesis. As an example, the full characterization of the product prepared by using a FRR = 8, hereafter RD-SCO-1(FRR8), is reported as Supplementary information (Figures S12-S21). Note that FRRs values lower than 2 or higher than 8 led either to flow instabilities or to fouling of the microfluidic channels due to extend crystallization in chip, and thus could not be explored. Also, in the case of RD-SCO-1(FRR8) sub-30 nm nanoparticles were obtained as confirmed by SEM, TEM and DLS analyses, with a qualitatively similar spin transition behavior. However, the chemical analyses showed a triazole/triazolate ratio of 2.3:0.7 (found chemical formula: [Fe(Htrz)2.3(Trz)0.7](BF4)1.3), i.e. with an increased number of charge defects than in the case of RD-SCO-1 prepared at FRR = 2 (Tables S3). This difference in the number of defects, albeit small, results in a slightly narrower hysteresis of the annealed form of RD-SCO-1(FRR8), i.e. a spin transition with Tc↑ = 336 K and Tc↓ = 320 K was observed (compare Table 1 and Table S4).
3. Conclusions
In conclusion, we have shown that a controlled interplay of a coordination reaction with mass transport is crucial not only to favor the isolation of the otherwise inaccessible kinetic product, but to control defect engineering. Herein, a prototype functional coordination polymer (SCO-1) was chosen as the best model compound to showcase the impact of RD conditions on materials properties. The data obtained clearly show that controlled defect engineering and RD microfluidic environments may be the only effective way of tuning structure-properties correlations in a systematic manner. Considering the growing interest in tuning and understanding the SCO phenomenon, and in general the physicochemical properties of porous and non-porous CPs, we believe that the presented approach will open new avenues towards unprecedented and unique functional materials and devices.
4. Experimental
4.1. Materials and methods
The reagents iron(II) tetrafluoroborate hexahydrate (Fe(BF4)2•6H2O) and 1,2,4-Triazole (C2H3N3) were purchased from Sigma-Aldrich GmbH (Germany) and used without further purification. HPLC grade ethanol was obtained from Fluka Chemie AG (Switzerland).
4.2. Microfluidic device fabrication
The silicon master mold used for fabrication of the microfluidic device employed in this work was fabricated through standard photolithographyprocesses extensively described elsewhere. [36] The microfluidic device was fabricated by replica molding of PDMS (SYLGARD 184 Silicone Elastomer Kit) against the previously fabricated master mold. In brief, a mixture of PDMS elastomer and curing agent (10:0.9 ratio by weight, respectively) was casted against the master mold, cured and cut to the target size. The inlets and outlet holes were punched using Biopsy punchers. Before bonding the device to a glass coverslip (Menzel-Glaser, Germany), a thin layer PDMS was spin-coated onto the glass coverslip. Using a corona discharge (Electro-Technic Products, USA) the PDMS slab (incorporating the microfluidic channels and inlet and outlet holes) was bonded to the PDMS coated glass coverslip, yielding the final device. Eventually, the fabricated microfluidic device comprised four inlet channels converging into a single one 1 cm long. The cross sectional dimensions of inlet channels are 50 µm × 50 µm and 250 µm × 50 µm for the 1 cm long microchannel (i.e. the microchannel where the reaction is conducted).
4.3. Microfluidic synthesis of RD-SCO-1
The reagent flows were injected into the inlets of the microfluidic device using a syringe pump system (neMESYS module, Cetoni GmbH, Germany). Ethanol solutions of Fe(BF4)2•6H2O (1M) and 1,2,4-triazole (3M) were injected into two middle inlets at the flow rate of 50 µL/min, while pure ethanol flows were injected into the two side inlets at a flow rate of 100 or 400 µL/min, for RD-SCO-1 (FRR2) and RD-SCO-1 (FRR8), respectively. The reaction mixture coming out from the outlet was collected in a glass bottle containing 100 mL pure ethanol during 10 min in order to quench by dilution the reaction after microfluidic synthesis.
4.4. Isolation of RD-SCO-1 powder
The suspension collected in the glass bottle was transfered in falcon tubes and centrifuged at 3000 rpm for 5 min in three consecutive steps. After each step, the supernatant was discarded from top and the remaining powder was re-suspended again in fresh ethanol. After three washing steps the suspension was poured onto a glass petri-dish and dried under vacuum yielding RD-SCO-1 as a pink powder (Figure S1).
4.5. Bulk synthesis of bSCO-1
Bulk SCO-1 compound was synthesized following the procedure reported in the literature. [16] A solution of 1,2,4-triazole (1.040 g, 1.5 × 10−2 mol) in EtOH (5 mL) was added to a solution of Fe(BF4)2•6H2O (1.687 g, 5 × 10−3 mol) in ethanol (10 mL). The purple suspension obtained was left undisturbed for 24 h and filtered. The resulting purple powder (bSCO-1) was washed with ethanol and dried in air overnight.
4.6. Scanning electron microscopy (SEM)
Samples of RD-SCO-1 and bSCO-1 for SEM measurements were prepared by drop casting a suspension of the solid material previously re-dispersed in ethanol on aluminum stubs followed by evaporation of the solvent under ambient conditions. Before performing the analysis, the samples were metalized with a 5 nm Pt coating.
4.7. Transmission electron microscopy (TEM)
A drop of the sample containing suspended RD-SCO-1 nanoparticles coming out from the outlet of microfluidic chip was poured on a TEM Grid (200 mesh, gold-based holey carbon film, Quantifoil®) in order to measure TEM of samples directly after the microfluidic reaction. Alternatively, previously isolated RD-SCO-1 powder was re-dispersed in ethanol and the obtained suspension drop cast onto TEM grids (ultrathin carbon type-A, 400 mesh Cu grids. Ted Pella Inc., Redding, CA, USA). In both cases, samples were dried overnight prior to examination.
4.8. Dynamic light scattering spectroscopy (DLS)
To perform the size distribution and Zeta-potential measurements, previously isolated RD-SCO-1 powder was re-dispersed in water or ethanol and 1 mL of re-dispersed sample was introduced in a disposable capillary cell.
4.9. UV–visible and ft-ir spectroscopy
Absorbance spectra for RD-SCO-1 samples were recorded by using a spectrophotometer in the visible- near-infrared range. The UV–vis and FT-IR measurements were performed on the same sample in the form of pellets. The temperature dependent measurements were done in the range of 298 K to 440 K.
4.10. Magnetic susceptibilities (SQUID)
Magnetic susceptibility measurements were performed on polycrystalline samples of RD-SCO-1 and bSCO-1. Data were corrected for diamagnetic contributions, which were estimated from the Pascal constants.
4.11. Inductively coupled plasma mass spectrometry (ICP-MS)
ICP-MS was used for the determination of the iron percentage in RD-SCO-1(FRR2), RD-SCO-1(FRR8) and bSCO-1 samples. For the quantification of metal, a dilution 1:100 was necessary for detecting the correct signals. The isotope 57Fe was selected as tracer. The iron concentration of each sample was measured using a calibration curve obtained in the range of 0.01−250 ppb of metal. Standard reference material (PerkinElmer) with known values of iron and rare earth elements was analyzed with each batch of samples. All the samples were measured per quadruplicate.
4.12. Thermogravimetric analysis
Thermogravimetric analyses (TGA) of samples were run with samples held in aluminum crucibles at the heating rate of 5 °C/min from 25 to 600 °C.
4.13. Small angle and wide angle X-Ray scattering measurements (SAXS/WAXS)
Transmission SAXS and WAXS were measured in a S3-MICRO (Hecus X-ray systems GMBH Graz, Austria) incorporating a X-ray source GENIX-Fox 3D X-ray source (Xenocs, Grenoble), which provides a detector focused beam with λ = 0.1542 nm Cu Kα-line (with more than 97% purity and less than 0.3% Kβ). Both detectors are PSD 50 Hecus. Temperature was controlled by means of a Peltier TCCS-3 Hecus or a resistance TCCS-300. The samples were inserted into glass capillaries with diameters of 1 mm or 0.2 mm and 10 μm wall thickness. The thinner capillaries providing improved signal in the WAXS region due to smaller absortion. The SAXS scattering curves are shown as a function of the scattering vector modulus:(1)where, 2θ is the scattering angle. The q values with this setup ranged from 0.08 nm−1 to 6.0 nm−1 in the SAXS regime (0.1° < 2θ < 8.5°). WAXS curves are shown as a function of 2θ with a range from 18.5° to 27° (13.1 to 19 nm−1 in q scale). The silver behenate and para-bromobenzoic acid were used to calibrate SAXS and WAXS measurements, respectively. To avoid the detector-width smearing effect, a 1 mm slit was used for the SAXS measurements. To improve the contact of the capillary with the Peltier Block, 1 mm capillaries were inserted in a brass tube with a notch to let X-ray pass. Instead, 0.2 mm capillaries were measured close to the tip of the brass tube also to ensure the good temperature transmission. Spectra were acquired in 20 min frames to complete three hour, then the temperature was raised to the high temperature and let to stabilize for 20 min prior to the acquisition. Then, the samples were cooled down to the low temperature. Each annealing cycle was conducted during three hours. Additionally, SAXS data was fitted to a polydisperse sphere model using a Schultz distribution.
4.14. Differential scanning calorimetry (DSC)
DSC measurements were carried out in a He atmosphere. The heating and cooling rates were fixed at 10 K/min. Temperatures and enthalpies were calibrated over the temperature range of interest (260–420 K) using the solid–liquid transitions of pure indium (99.99%). The DSC data successively recorded several times for all samples and the evaluated enthalpy and entropy variations are reported in Table S2.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author Statement
D.R. and J.P.L. conceived the idea and led the project; A.A., S.S-G. and S.S. conducted the experiments and analyzed the data. A.A. and S.S. fabricated the microfluidic devices; A.S. helped to interpret the data and contributed to the development of the manuscript idea and to its writing; R.P. performed and interpreted the SAXS data; all authors wrote and revised the manuscript.
Acknowledgement
This work was partially supported by the European Union (ERC-2015-STG microCrysFact 677020), the Swiss National Science Foundation (Projects no. 200021_160174 and 200021 181988), MAT 2015–70615-R and MINECO CTQ2017-88948-P and RTI2018-098027-B-C21 from the Spanish Government funds, and by the European Regional Development Fund (ERDF). J.P.L. and S.P. acknowledge networking support by the COST Action CM1407 “e-minds”. The ICN2 is funded by the CERCA programme / Generalitat de Catalunya. The ICN2 is supported by the Severo Ochoa Centres of Excellence programme, funded by the Spanish Research Agency (AEI, grant no. SEV-2017-0706). S.S-G. acknowledges the support from MINECO BES-2015-071492 grant. A.R. acknowledges the European Commission through the SPINSWITCH project (H2020-MSCA-RISE-2016, Grant Agreement No. 734322). Additionally, all authors acknowledge Jaume Caelles for SAXS/WAXS measurements performed at IQACCSIC. G.A. thanks support by the Generalitat de Catalunya through the ICREA Academia Prize 2018 Authors A. Abrishamkar, S. Suárez–García and S. Sevim contributed equally to this work.
CTQ2017-88948-P and RTI2018-098027-B-C21 from the Spanish Government funds, and by the European Regional Development Fund (ERDF). J.P.L. and S.P. acknowledge networking support by the COST Action CM1407 “e-minds”. The ICN2 is funded by the CERCA programme / Generalitat de Catalunya. The ICN2 is supported by the Severo Ochoa Centres of Excellence programme, funded by the Spanish Research Agency (AEI, grant no. SEV-2017-0706). S.S-G. acknowledges the support from MINECO BES-2015-071492 grant. A.R. acknowledges the European Commission through the SPINSWITCH project (H2020-MSCA-RISE-2016, Grant Agreement No. 734322). Additionally, all authors acknowledge Jaume Caelles for SAXS/WAXS measurements performed at IQACCSIC. G.A. thanks support by the Generalitat de Catalunya through the ICREA Academia Prize 2018 Authors A. Abrishamkar, S. Suárez–García and S. Sevim contributed equally to this work.