1. Introduction
Polyamides (PAs) are engineering thermoplastics with a broad range of applications. Most of the PAs are synthesized in polycondensation processes resulting in versatile polymeric backbones due to the big variety of diamine and dicarboxyclic acid compounds. PAs, containing the representative amide groups ( CONH), are either amorphous or semicrystalline. Beside of polycondensation, some PAs can be synthesized by anionic ring-opening polymerization (AROP) of the corresponding cyclic lactams. The number of the technologically relevant AROP-prone lactams is limited, however, for ε-caprolactam (CL – as precursor of PA6) and ω-laurolactam (lauryllactam) (LL – as precursor of P-A12). The first description of the AROP of CL is dated back to 1941 [1], [2]. Since then a large body of works was dealing with different aspects of the AROP, tailoring the properties, broadening the processing, and enhancing the production effectiveness of in situ polymerized PA6 and PA12.
CONH), are either amorphous or semicrystalline. Beside of polycondensation, some PAs can be synthesized by anionic ring-opening polymerization (AROP) of the corresponding cyclic lactams. The number of the technologically relevant AROP-prone lactams is limited, however, for ε-caprolactam (CL – as precursor of PA6) and ω-laurolactam (lauryllactam) (LL – as precursor of P-A12). The first description of the AROP of CL is dated back to 1941 [1], [2]. Since then a large body of works was dealing with different aspects of the AROP, tailoring the properties, broadening the processing, and enhancing the production effectiveness of in situ polymerized PA6 and PA12.
Next, we shall survey the most important developments whereby considering their industrial potentials. Owing to this treatise, the reader may miss some aspects which were classified by us as industrially less relevant and thus not, or only tangentially mentioned in this work. On the other hand, the eventually missing information can be gathered from excellent books, book chapters and reviews on the field of AROP of lactams [1], [3], [4], [5], [6], [7].
2. AROP of lactams – chemistry
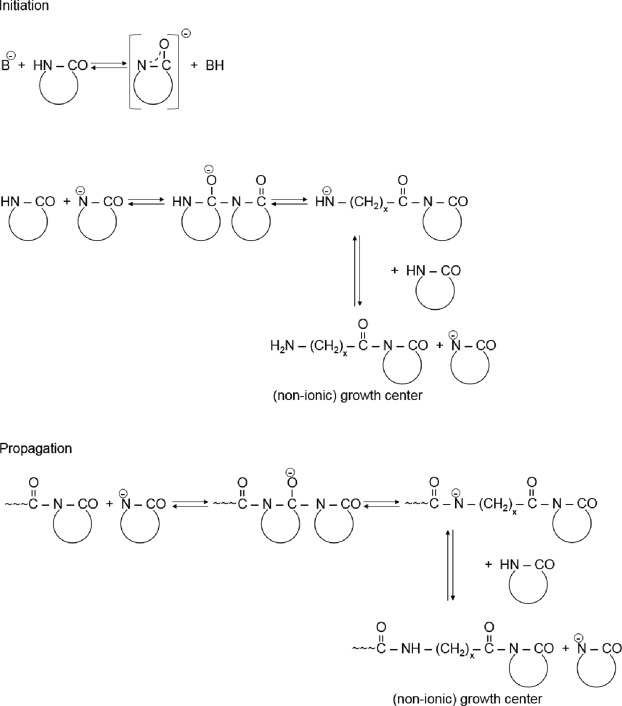
Though the in situ polymerization of lactams can be initiated by acids (cationic polymerization), it is seldom used [8], because it yields low molecular weight(MW) products at low conversions [1], [9]. Nevertheless, recent interest in the academic research for the cationic polymerization of lactams is fueled by the possible intercalation of lactam onium cations between the graphite layers [10]and good dispersion of other carbonaceous nanofillers, such as fullerenes [11]. The technologically relevant polymerization of lactams is initiated by strong bases, and thus termed as to anionic polymerization. The mechanism of the AROP of lactams, summarized in the 1970s [1], [12], [13], is well known. Accordingly, strong bases, forming the free lactam anions, work as the initiators (see Fig. 1). Because the amine (aminic) anion is more basic than the initial lactam anion, a new lactam anion and ω-aminoacyllactam form via fast proton exchange. Propagation proceeds by repeated nucleophilic attack of the lactam anion on the endocyclic carbonyl group of the non-ionic growth center. Thus, the propagation consists of repeated acylations of the lactam anions (cf. Fig. 1).
 Fig. 1. Initiation and propagation of the AROP of lactams, schematically. Note: x = 5 and = 11 for CL and LL, respectively.
Fig. 1. Initiation and propagation of the AROP of lactams, schematically. Note: x = 5 and = 11 for CL and LL, respectively.The formation of N-acylated lactam is the controlling step of the non-activated ROP of lactams. It is therefore intuitive that the propagation rate of AROP can be prominently enhanced by the introduction or in situ generation of compounds of N-acyllactam structure which may overtake the role of the growth centers (see Fig. 1). They are called activators in this work though they are termed – similar to the initiators – differently in both the open and patent literature. The in situ polymerization in presence of activators is referred to activated anionic (or fast) polymerization for which we shall use further on AROP because an activator is practically always present in the corresponding formulation.
The initiators, produced ex situ or in situ, are mostly alkaline salts of lactams. Sodium caprolactamate (NaCL), by whatever method produced, is the most widespread initiator. NaCL is commercially available under the trade names Bruggolen® C10 (C10, Brüggemann Chemical, Heilbronn, Germany) and Addonyl® CR CATALYZE and Addonyl® Kat NL (Rhein Chemie of Lanxess, Cologne, Germany) in which NaCL is dissolved in CL (“masterbatch”). Another commercial initiator is the ε-caprolactam magnesium bromide (CLMgBr), offered as Nyrim® C1 (C1, also in masterbatch form) by Brüggemann Chemicals. A further commercial, but solvent-based, initiator (Dilactamate®) is sodium dicaprolactamato-bis-(2-methoxyethoxo)-aluminate, produced by Katchem (Prague, Czech Republic).
The activators are classified as direct or indirect ones. Direct activators, exhibiting an N-acyllactam structure are produced ex situ, while compounds yielding such structures in situ through chemical reactions are considered as indirect ones [1]. In the academic research a great number of different direct and indirect activators have been used and the related research is still ongoing. Many recent attempts try to ensure the activators with other functionalities, such as flame retardant effect [14], or enhanced branching/crosslinking [15], [16]. Enhanced melt strength guaranteed by branching, partial crosslinking is a key parameter for the in situ production of foams. Nevertheless, for the CL polymerization N-acetyl-ε-caprolactam and various diisocyanates are the preferred direct and indirect activators. Nowadays, the industrial available activators are of N-carbamoyllactam types. Bruggolen® C20 (C20) for example contains hexamethylene-1,6-dicarbamoylcaprolactam as active species in CL masterbatch. The active substance in Addonyl® ACTIVATE is a CL-blocked isocyanate. N-carbamoyllactam precursor polyisocyanates, as indirect activators, are offered by Rhein-Chemie (Lanxess) under the trade names Addonyl® 8101 and Addonyl® TT. Polycarbodiimide-based activator has also been commercialized (Addonyl® P). The growth centers from carbodiimide are formed through the reaction with NaCL yielding a guanidine structure [1]. The carbodiimide activator was the essential part of the initiator/activator package (Grilonit LA), developed by Ems Chemie (Domat, Switzerland), for the anionic LL polymerization [17]. A further activator with industrial perspectives are N,N′-alkylene bisamides [18], including N,N′-ethylene-bisstearamide [19].
This is the right place to call the attention to some key issues with respect of AROP of CL and LL. First, the initiator and activator, each dissolved in the corresponding lactam (masterbatch), are introduced separately into the molten lactam to be polymerized. Second, the AROP is extremely sensitive the moisture (water is an inhibitor of the polymerization [1], [9]) and thus the polymerization has to be carried out under inert atmosphere (nitrogen is preferred) or under vacuum. Nonetheless, in some cases (e.g. LL polymerization) researchers did not make any precaution (e.g. drying, inert gas blanketing) arguing that at the polymerization temperature set (in case of LL T ≥ 150 °C), the residual moisture is fully removed [20]. The sensitivity to moisture of the AROP of lactams remains most likely a critical issue in the future. On the other hand, the reader may be surprised by the fact, that less work is available on how the residual moisture content affects the polymerization (degree of conversion, DOC) and MW characteristics of the final product. The separate introduction of initiator and activator represents a “handling” problem for the industry, therefore attempts were already made for their common dosage. The Grilonit LA (no more commercialized) of Ems Chemie contained NaCL initiator and a carbodiimide (cycloaliphatic monocarbodiimide) in a solvent (dimethylpropylene urea). Because neither the initiator nor the activator was in contact with the lactam, the shelf life of this initiator/activator compound was practically indefinite [20], [21]. Further common feature of the AROP of CL and LL that their polymerization temperatures should lay above the melting temperature (Tm) of the corresponding lactam, where the polymerization reaction is already rather fast (see later). So, the question arises, whether or not is it possible to initiate the polymerization “on demand”? This requires the use of latent initiator/activator systems. Though research work addressed already the development of thermally activated latent initiators, the DOC and MW data are still far from the expected and industrially relevant values [22], [23].
3. Polymerization and properties
The anionic polymerization of CL and LL can be carried out in two temperature ranges: below or above the melting temperature (Tm) of the resulting PAs. Note that Tm of PA6 and PA12 lay in the temperature ranges 210–225 °C and 180–190 °C, respectively. The polymerization temperature affects the DOC, MWvalues, crystallinity and thus also the overall thermal and mechanical propertiesof the resulting PAs. As it will be shown later, AROP below Tm is strongly favored for industrial applications. This is because polymerization is accompanied with crystallization and thus the solidified final part can be earlier demolded compared to the polymerization above Tm where additional cooling is required for the crystallization (solidification). On the other hand, polymerization above Tm is necessary when AROP is used for the in situ preparation of homo-, copolymers and blends for further processing, such as injection molding, fiber spinning etc.
The DOC of CL and LL to the corresponding linear PA6 and PA12 polymers, respectively, can be followed by different techniques. The residual amount of CL in PA6 for example can be determined by extraction (water, methanol), drying in vacuum or via thermogravimetric analysis (TGA) [24]. The two latter techniques make use of the fact that CL easily evaporates (boiling temperature of CL is below that of the onset of the thermal degradation of PA6).
A more advanced but still not frequently practiced method is the gel permeation chromatography (GPC) that informs us also about the MW characteristics (number- and weight average data, Mn and Mw, respectively) and polydispersity (Mw/Mn) [25]. On the other hand, the most widely used technique is the solution viscosimetry yielding the viscosity-average MW (Mv). From the intrinsic viscosity values, measured in diluted solutions of suitable solvents (sulfuric acid, formic acid, cresols [13]), the Mv values are computed through the Mark-Houwink–Sakurada equation [26] (note this equation is differently credited to its proposers but Mark–Houwink is usually involved). The MW of PA6 and PA12 may reach ca. 100 kDa [19] though the usual MW range is 20–40 kDa which agrees fairly with those of commercial PA6 and PA12, manufactured by the hydrolytic route.
Polymerization below Tm is favored as it yields high conversion (DOC = 96–99%) and high crystallinity (40–50%). Note that PAs crystallized from the melt show lower crystallinities, viz. in the range of (30–40%) [1]. Recall that the most beneficial behavior of the AROP below Tm is that it is accompanied with fast solidification owing to crystallization.
Irrespective whether crystallization takes place after or simultaneously with the polymerization, it occurs in an undercooled melt. The occurrence of polymerization and crystallization has been topic of intensive research. Karger-Kocsis et al. [27], [28] concluded based on differential scanning calorimetry(DSC) results that polymerization and crystallization are superimposed processes in the AROP of CL that was confirmed also in a very recent paper [29]. Based on a detailed DSC study Vicard et al. [30] concluded that at high temperatures (isothermal DSC scans) or high heating rates (dynamic DSC scans) polymerization precedes the crystallization because of the small extent of undercooling. Under given conditions the polymerization may be even fully decoupled from the crystallization. The complete separation of the polymerization and crystallization allows us to determine the total enthalpy of polymerization, as well [30]. Note that with enlargement of the cyclic rings the reaction enthalpy is usually reduced. This is the case when one compares the related data for CL [30] and LL [21], respectively. Important aspect is, however, that AROP of CL and LL is exothermic, and crystallization is also an exothermic process, which should be considered during processing accordingly.
A peculiar feature of the AROP of lactams is that it represents a suitable method to produce PAs in powder form [1]. To produce powders, the polymerization is carried out in suitable organic liquids acting as precipitants. The size of the powders may be micron- [31] or even nanoscale [30]. Such powders with or without further nanoadditives (see later) may reach an industrial breakthrough in the manufacturing of composites. Composites can be easily produced from powder-impregnated reinforcements of various types via various molding operations, such as compression molding, pultrusion [32], [33]. Note that in this case the reinforcement is wet-out and infiltrated by the molten PA derived from the powder.
The powders themselves can be preferably used for coatings (flame spraying, electrostatic coating), binders [34] and carrier materials in different fields [35]. Porous PAs, exhibiting extreme high crystallinities (reaching 60%), can also be produced via AROP in presence of suitable solvents, as demonstrated recently by Rahman et al. [34].
Both PA6 and PA12 are semicrystalline, polymorphic polymers. Each of them have two basic crystalline structures such as α monoclinic and ϒ (pseudo)hexagonal [13]. Their appearance depends not only on the synthesis and processing conditions but also on other factors, like presence of additives, fillers.
The fact that the polymerization of CL can be adequately described by the Kamal–Sourour equation [36], [37], [38], developed for thermoset curing, already hints for some similarities between the in situ polymerization and curing of thermoset resins. In both cases the viscosity (chemorheology) and conversion increase as a function of time and temperature.
For thermosets the above changes are summarized in conversion-temperature-transformation (CTT) and time-temperature-transformation (TTT) diagrams. These diagrams are of great practical relevance being suitable tools optimize the curing process. Knowing the changes of the AROP of lactams in time/temperature scales is a key factor for their industrial implementation. Pioneering work in this field was done by Luisier et al. [21] who constructed a TTT diagram for the AROP of LL. The input parameters to this diagram (conversion, crystallinity) were derived from isothermal and dynamic DSC scans. The related TTT diagram indicated the changes in the DOC and onset of crystallization at different polymerization temperatures (both below and above the Tm of PA12).
The recent work of Maazouz et al. [39] put emphasis on the chemorheology, i.e. viscosity change as a function of both polymerization temperature and time, when creating a TTT diagram for the AROP of CL. The authors assessed the conversion by Fourier-transform infrared spectroscopy (FTIR) and measured the melt viscosity, as well. Moreover, dielectric spectroscopy measurements were tried to follow the course of polymerization. The TTT diagram of Maazouz et al. [39] is of great practical relevance because it informs us about the presence of a viscosity range lower than 1 Pa s. It has to be underlined that 1 Pa s is generally considered as the maximum threshold of all “reactive” resins used in resin transfer molding (RTM) or similar operations.
4. Modifications
Usual targets of the property modifications of AROP-produced PAs are: reduced crystallinity, lowered melting temperature, enhancement of ductility and toughness, increase of the resistance to thermal and hydrothermal degradations. Basic strategies to reach the above improvements are: in situcopolymerization, blending and (nano)filling.
Copolymerization of CL with LL, being industrially available lactams, is one of the most straightforward approach. The related formulations usually contain more CL than LL because CL is more reactive that LL. Based on the topic-related excellent summary of Roda [1], the AROP of CL and LL with NaCL initiator yielded random copolymers exhibiting low Tm (minimum value ∼135 °C) and reduced crystallinity compared to PA6. In contrast, copolymers synthesized with CLMgBr initiator displayed two distinct Tms (in the ranges T = 130–140 °C and T = 200–220 °C, respectively) suggesting the formation of block copolymer, when the actual CL content was between 30 and 70 mol %. Kinetic investigations confirmed that in the initial stage of polymerization PA6 homopolymer forms while random chains, consisting of CL and LL, appear in a later stage [1], [40]. Rusu et al. [41] studied the activated anionic copolymerization of CL and LL in centrifugal molding using CLMgBr initiator and N,N′- isophtaloyl-bis-ε-caprolactam activator at 160 °C. The LL amount in the feed was varied between 0 and 50 wt%. It was found that the conversion, Mv, degree of crystallinity all were reduced with increasing LL content. Parallel to that, however, the flexural modulus and water uptake decreased while the Izod impact strength enhanced. The above listed selected results underline that the copolymerization of CL and LL is a very complex process. The relative amount of the comonomer and its position along the polymer chain depend on several factors (reactivity of each activated monomer, acylation of the lactam anions, possible side reactions, crystallization reactions etc.). Lactones may also be suitable comonomers. Note that they act as activators in the AROP of lactams until their amount remains below 5 mol% [1]. In this case the initiation step is the acylation of lactone by the lactam anion [1], [42]. At higher concentrations, lactone may work as activator and comonomer at the same time. It should be kept in mind that copolymerization is a useful tool to reduce the crystallinity, lower the Tm, to reduce the water uptake and to enhance the ductility.
On the other hand, the right tool for ductility and toughness improvements is the synthesis of block copolymers. The preparation of copolymers with blocks composed of lactam or non-lactam chains requires the incorporation of suitable prepolymers in the molten, anionically polymerizable lactam. Such prepolymers should be soluble in the lactam melt and, in addition, bear reactive end groups, such as hydroxyl (OH; e.g. polyether- or polyester-type diols) and amine (-NH2; e.g. polyether diamines). The key step of the block copolymerization is to transfer the above end groups to N-acyllactam or N-carbamoyllactam moieties. Afterward, they function as so called macroactivators in the subsequent AROP of CL or LL. The related “transformation precursors” are polyisocyanates (yielding N-carbamoyllactam) or bis-acyllactams which undergo exchange reactions (alcoholysis and aminolysis with diols and diamines, respectively) [1]. Poly (ε-caprolactam-block-polyether) copolymers were produced, and the related components offered commercially by Monsanto, in 1982 (Nyrim® technology). The related block copolymers are referred to as nylon (also called Nyrim) block copolymers (NBC) or nylon reaction injection molding (Nylon RIM) products. The Nyrim® activity was terminated in 1985 by Monsanto and sold to DSM afterward. It is now commercialized by Brüggemann Chemicals.
Nylon RIM (or NBC) block copolymers are obtained in the AROP of CL in the presence of polyols (polyether- or polyester-types) and bis-acyl derivatives of CL [43] – see Fig. 2. The initiators are NaCL, and more frequently CLMgBr. Note that the water uptake of NBC is lower and the toughness is prominently higher than those of the reference PA6.
 Fig. 2. Scheme of the preparation of poly(amide-block-ether) or poly(amide-block-ester) according to the Nyrim® technology.
Fig. 2. Scheme of the preparation of poly(amide-block-ether) or poly(amide-block-ester) according to the Nyrim® technology.Research in the academia demonstrated the possibility of incorporation of other block segments into NBCs than polyesters or polyethers [44], [45], [46]. Due to the industrial availability of polyether- and polyester-type diols and diamines, which are widely used in the polyurethane synthesis and for epoxy hardening, respectively, these modifiers remain in the forefront of applications [47], [48], [49], [50], [51].
The successful and broad applications of thermoplastics is due to their versatile blending. Therefore, it is not surprising that in situ grafting and blending of/with various thermoplastic polymers making use of the possibilities offered by the AROP of lactams was explored in the academic research [52]. The target properties were as mentioned before, i.e. reduction of water uptake, increasing thermal and hydrothermal resistance, and particularly, improving the toughness. The prerequisite of this modification strategy is that the polymer modifier should be dissolved in the molten lactam prior to the polymerization of the latter. Note that lactams are efficient solvent for many polymers. Polymers, such as polystyrene [53], [54], styrene-acrylonitrile copolymer [55]acrylonitrile-butadiene rubber [56] can be easily dissolved in molten lactams prior to their AROP. Blending with the in situ AROP of lactams is a very promising route, because the morphology development becomes instead of thermodynamically mostly kinetically controlled. A further tool of the morphology control of in situ produced blends or grafted copolymers is to use functional (such as thermoplastic polyurethane [57]) or functionalized polymers (such as hydroxyl- or isocyanate end-capped ethylene-butylene elastomer [58], acrylate compound grafted polyphenylene oxide [59]). Note that in the latter cases the blend component itself acts as macroactivator of the AROP. This kind of blending (grafting) process is then straightforward when the final part is manufactured. e.g. composite parts with in situ polymerized matrices as graft copolymers or blends. Interestingly, this aspect was not yet addressed even by the academic research. On the other hand, the industrial relevance of blends synthesized through in situ AROP of lactams is questionable when reprocessing (e.g. via injection or extrusion molding) of the grafted copolymers and blends is foreseen. This claim is reasoned by the fact that the morphology is unstable and thus may markedly change upon remelting.
Among possible property modifiers special attention has to be paid to the nanofillers [52]. The nanofillers, -reinforcement can be produced ex situ(preformed particles) or in situ (sol-gel route, intercalation/exfoliation with/in the corresponding polymer). The nanofillers, having at least nanoscaledimension in one space direction, are usually grouped into (quasi)spherical, needle-like, and platelet-like types. These groups are frequently termed to 0 direction (0D), 1D and 2D nanofillers, respectively. Their incorporation into PA6 and PA12 via AROP may substantially increase the stiffness, strength, heat distortion temperature and resistance to thermal degradation. Since many nanofillers are of polar character and bear polar functional groups, such as -OH, -COOH, their surface should be rendered organophilic. AROP of lactams offers interesting possibilities for surface grafting. The strategy here is exactly the same what was mentioned with respect to the in situ graft copolymerizationand blending: generation of a (macro)activator for the subsequent AROP of lactam. For example, the hydroxyl groups on the filler's surface can be transformed into N-acetyl or N-carbamoyl groups via suitable chemistry thereby producing the activator sites for AROP. This approach is referred to as “grafting from” since in this case grafting of the PA chain starts from the surface of the nanofiller – see Fig. 3.
 Fig. 3. Synthesis of PA6/silica nanocomposite in AROP via the “grafting from” approach. Designations: 1 – toluene -2,4-diisocyanate (TDI), 2 – CL-capping of the free isocyanate to create the macroactivator, 3 – AROP of CL when initiated by the traditional way ([60] reproduced with the permission from BME PT).
Fig. 3. Synthesis of PA6/silica nanocomposite in AROP via the “grafting from” approach. Designations: 1 – toluene -2,4-diisocyanate (TDI), 2 – CL-capping of the free isocyanate to create the macroactivator, 3 – AROP of CL when initiated by the traditional way ([60] reproduced with the permission from BME PT).As 0D nanofillers mostly silica [61], [62] and metal-oxides [63] have been tried as property modifiers. Their incorporation markedly enhanced the heat distortion temperature, stiffness and strength, however the toughness usually went through a maximum as a function of filler loading owing to agglomeration phenomena. Incorporation of micron scaled graphite with and without coupling agent bearing activator sites reduced the coefficient of friction and wear rate [64], [65]. The latter is of great practical relevance as cast PAs are generally used in tribological applications. A similar effect may be expected from fullerenes, the effect of which was however tested for electrical conductivity [66], [67]. In case of the 1D multiwall carbon nanotubes (MWCNT) the grafting from approach was adopted [68], [69], [70], [71]. Therefore, the CNTs were previously functionalized by oxidation before transforming them into “macroactivators” using polyisocyanates. As expected, the incorporation of MWCNT enhanced the stiffness and strength, usually at cost of the ductility (especially when in higher amount applied). Similar results were reported for cellulose nanocrystals which might have participated in the formation of a strong interphase via transamidation reactions [72]. Research on 2D nanofillers focused on clays (with and without organophilic modification) and graphenes. Their incorporation strongly enhanced the stiffness but the strength changed as function of the intercalation/exfoliation states [52]. Similar to MWCNT, graphenes delayed the polymerization and reduced in some extent the MW. On the other hand, the 2D nanofillers strongly enhanced the resistance to thermal degradation [73], [74]. Changes in the polymorphism was mostly reported for 2D-type nanofillers [75]. Improvement in the barrier performance can be expected from 2D nanofillers which can be exploited in items produced by centrifugal or rotational molding.
Solvent-assisted AROP of lactams may be a straightforward strategy to produce nanocomposites in powder form as demonstrated in a series of papers by the group of Denchev [76], [77], [78]. This micro-capsulation route is outlined along with the related chemistry in Fig. 4. Note that bulk nanocomposites can be produced from the powder by sintering, compression or injection molding. Moreover, the nanocomposite powder may serve as matrix-forming precursor in composites with traditional fiber/fabric reinforcements (see later).
 Fig. 4. Morphology development of the nanocomposite during in situ solvent-assisted micro-capsulation (top) and the related chemistry (bottom). Designations: AAP – activated anionic polymerization, MP – melt processing above Tm of PA6, C20 – Bruggolen C20 activator, DL –Dilactamate® – dicaprolactamo-bis-(2-methoxyethoxo)-aluminate, ECL – ε-CL ([76] reproduced with the permission from BME PT).
Fig. 4. Morphology development of the nanocomposite during in situ solvent-assisted micro-capsulation (top) and the related chemistry (bottom). Designations: AAP – activated anionic polymerization, MP – melt processing above Tm of PA6, C20 – Bruggolen C20 activator, DL –Dilactamate® – dicaprolactamo-bis-(2-methoxyethoxo)-aluminate, ECL – ε-CL ([76] reproduced with the permission from BME PT).It is the right place to underline that the knowledge acquired by the academic research in the field of in situ AROP-produced nanocomposites has not been transferred to the industry.
5. Processing and applications
In this chapter we shall summarize relevant developments from the sites of processing (traditional and novel techniques) and materials. Although reactive extrusion of lactams, lactam copolymers and blends were in depth investigated in the academia [79], [80], [81], [82], [83], and its feasibility unequivocally demonstrated, this topic remains without industrial interest, and thus will not be treated.
5.1. Casting
Casting is the eldest processing method making use of the AROP of lactams. In this process the initiator- and activator containing molten lactams are mixed and poured into a preheated “open” mold. Though the usual products are sheets, rods, tubing, discs, billets and the like, also near net-shaped blanks can be produced. The low internal stress level (achieved through cautious cooling) in the cast products allows their subsequent machining. Cast PAs usually replace metallic parts and they are used for bearing, sheaves, pulleys, gears, rollers, sprockets and the like. In many applications low friction and high resistance to wear are the basic requirements. Important developments in this field are expected from the listed above material's modification possibilities. Accordingly, incorporation of carbonaceous nanofillers to improve the tribological properties and manufacturing of electric and heat conductive semi-finished products can be predicted. As far as tribological performance concerns the use of ionic liquids [84] as potential lubricants, may be promising. Reinforcement of in situ produced lactam polymers and related systems with discontinuous glass, carbon fibers is a promising route of property improvements [85]. Casting remains the basic tool and the first step of material development in the academic research.
5.2. Centrifugal and rotational molding
Centrifugal molding is used to produce finished or semi-finished parts of circular cross-sections. By contrast, via “reactive” rotational molding hollow bodies of very complex shapes can be manufactured. The attribute “reactive” refers to the fact that the related PA is produced in situ. Note that centrifugal and rotational molding operations can be performed also with previously polymerized PAs [86], available in their powder forms. The basic difference between reactive and traditional centrifugal and rotational molding is that in the former case polymerization can be carried out below, whereas in case of traditional molding only above Tm of the actual PA. As emphasized before polymerization below Tm, accompanied with crystallization (solidification) allows to shorten the cycle time which is of great industrial relevance. Centrifugal molding is seldom adapted in the academic research except the group of Rusu [41], [87]. It was reported that by proper selection of the polymerization temperature the stiffness, strength and toughness properties of the centrifugal molded items can be compromised. The control parameter behind is the crystallinity: lower crystallinity is accompanied with reduced stiffness and improved toughness [87]. Like to centrifugal molding, the proper processing parameters of rotational molding control the mechanical performance of the final product. Harkin-Jones and Crawford [88] investigated the influence of the initial mold temperature on the crystallinity degree and crystal size of rotation molded Nyrim® with and without short glass fiber (GF) reinforcement. They observed that crystallinity drops sharply when the mold temperature exceeds 140 °C. This finding agrees with that of deduced from centrifugal molding [87]. It was also reported that the incorporation of 5 mm length GF improved the stiffness and strength but at cost of the impact strength. Barhoumi et al. [89] investigated the AROP of CL during rotational molding. As initiators NaCL and CLMgBr, whereas as activator the bifunctional hexamethylene-1,6-dicarbamoylcaprolactam were selected. The polymerization performed at different temperatures below the Tm of PA6. It was found that induction time of the starting polymerization depended on the polymerization temperature, type and amount of the initiator/activator formulations. To support the reactive rotational molding operations of in situ polymerized PA6, the authors summarized the chemorheological results in TTT diagrams. Considering the fact that also for rotational molding the ideal viscosity should be less than 1 Pa·s, a suitable processing window (time available at a given temperature to reach this threshold viscosity) could be defined [89]. In the cited work also the properties of samples produced by traditional (fusing of PA6 powder/flake) and reactive rotomolding (AROP of CL), respectively, were compared. Table 1 clearly shows the benefits of the reactive rotational molding over the traditional one (based on ref. [89]).