1. Introduction
1.1. Trend of COVID-19
Recent outbreak of coronavirus disease 2019 (COVID-19) is attributable to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 has high sequence homology (shares 96.2% sequence identity) with the bat coronavirus RaTG13, implying the virus may have originated in bats and latterly crossed to humans in 2019 [1,2]. Parallel to severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV-2 presents as respiratory infection with symptoms of fever, cough, and pneumonia, but in severe cases may be progressing into organ failure [3].
With SARS-CoV-2 as an emergent human virus since December 2019, the world population is susceptible by reason of the deficiency in proper treatments for COVID-19. Generic or repurposed drug candidates are in trials, as yet with unremarkable results. Hydroxychloroquine, as an antimalarial drug, demonstrated antiviral activity against SARS-CoV-2 at micro molar concentrations in tissue culture, with clinical benefit in observational trials involving small number of patients; but showed no effect in reducing mortality for the large observational clinical trials [4], [5], [6]. Lopinavir-ritonavir, a human immunodeficiency (HIV)-1 protease inhibitor, demonstrated antiviral activity against SARS-CoV in tissue culture and infected patients; but showed no clinical benefit against SARS-CoV-2 [7]. Remdesivir, originally designed to treat Ebola virus infection, also displayed broad-spectrum antiviral activity against ribonucleic acid (RNA) viruses including SARS-CoV and MERS-CoV in tissue culture and animal models [8]. Remdesivir has been found to inhibit the RNA-dependent RNA polymerase of SARS-CoV-2, which improve recovery time in COVID-19 patients, nonetheless showed no effect in reducing mortality [9,10].
Active immunization has been considered, with multiple COVID-19 vaccines have been permitted under emergency use [11]. Sinovac's CoronaVac, as inactivated vaccine, utilizing SARS-CoV-2 which has been killed by physical or chemical ways to trigger an immune response. AstraZeneca/Oxford's AZD1222, as viral vector vaccine, utilizing non-replicating adenovirus as a vector containing genetic material of SARS-CoV-2 to trigger an immune response. Moderna's mRNA-1273, as RNA-based vaccine made from the viral sequence of SARS-CoV-2, with the immune cells processing the mRNA to manufacture protein that triggers an immune response [12]. Nevertheless, the timeline for developing a safe, effective and widely available vaccine for SARS-CoV-2 remains tentative. Besides, vaccine may not be 100% effective for immunocompromised individuals, therefore with therapeutics would be beneficial [13]. Passive immunization via the transfusion of serum collected from COVID-19 convalescent individuals to the critically ill COVID-19 patients has led to better clinical outcomes, suggesting neutralization of virus by the existing antibodies is useful [14]. To date, specifically designed neutralizing antibody therapies such as Regeneron's REGEN-COV (casirivimab with imdevimab), Lilly's bamlanivimab with etesevimab, GlaxoSmithKline and Vir Biotechnology's sotrovimab, AstraZeneca's Evusheld (tixagevimab with cilgavimab), Lilly's bebtelovimab have received authorization by the U.S. Food and Drug Administration (FDA) for emergency use in COVID-19 treatment [15], [16], [17].
SARS-CoV-2 was marked as one of the most transmissible coronaviruses that spreading rapidly and unceasingly throughout the world, adversely impacted human health while resulting in medical burden and lives lost. In March 2020, World Health Organization (WHO) declared COVID-19 as the first coronavirus pandemic in history, to be a public health emergency of international concern [18,19]. In course of prolonged infections, SARS-CoV-2 with escape mutants emerged. SARS-CoV-2 variants have been arisen in different countries due to the selection pressure across the worldwide spread, associated by the RNA virus nature with high mutation rate [20]. Till late 2021, five SARS-CoV-2 variants of concern have been identified: B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta), and B.1.1.529 (Omicron) [21], [22], [23]. Prevalent circulating variants of SARS-CoV-2 wane vaccine-elicited serologic responses and evade recognition and neutralization by clinical antibodies, as a daunting challenge that confront the development of therapeutics [24,25].
The pandemic caused by SARS-CoV-2 continues to spread, resulted in over 601.19 million infections and over 6.48 million deaths worldwide with the numbers still rising as of 2 September 2022 [26]. COVID-19 pandemic gradually leads to the collapse of healthcare systems, imposes a substantial social burden while causing tremendous economic losses worldwide. Hence, therapeutics that can stop as well as prevent SARS-CoV-2 infection in an effective manner are in crucial need. The development of therapeutics is actively in progress, with the recognition of therapeutic neutralizing antibodies as immediate and direct-acting antiviral agents accounted for short-to-medium term approach to combat COVID-19.
1.2. Characteristics of the cause for COVID-19: SARS-CoV-2
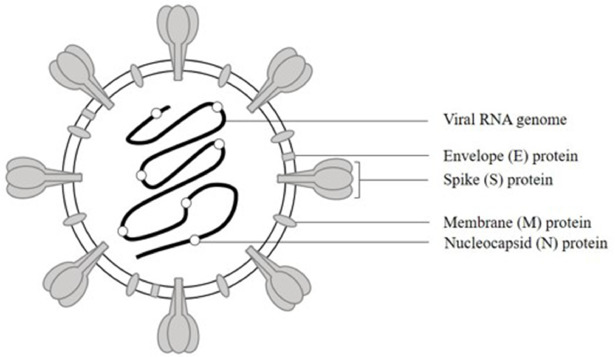
Coronaviruses can be categorized into four genera (α, β, γ, δ) which can infect a wide range of host organisms [27]. Thus far, seven types of coronaviruses that can cause disease in humans have been identified. HCoV-HKU1, HCoV-OC43, HCoV-NL63, HCoV-229E circulate seasonally and globally while causing mild respiratory disease [28]. SARS-CoV, MERS-CoV, and SARS-CoV-2 are zoonotic pathogens that have entered the human population over the last two decades, causing severe respiratory symptoms with high mortality and eventually leads to epidemics or pandemics [29], [30], [31]. SARS-CoV-2 which classified into the genus betacoronavirus in the family Coronaviridae, are more transmissible than previous coronaviruses [32,33]. In view of structural properties, SARS-CoV-2 is an enveloped virus, with positive-sense and single-stranded RNA genome (29,903 nucleotides in length) which encodes replicase and four major structural proteins: spike, envelope, membrane and nucleocapsid, like SARS-CoV [1,32,34]. Spike (S), envelope (E), and membrane (M) proteins are majorly incorporated into SARS-CoV-2 envelope lipid bilayer, enclosing a helical capsid formed by nucleocapsid (N) proteins bound to the RNA genome; hence formed the SARS-CoV-2 virion (Fig. 1) [1].
 Fig. 1. The structure of SARS-CoV-2 as a virion, including envelope (E), spike (S), and membrane (M) proteins incorporated into SARS-CoV-2 envelope, and nucleocapsid (N) protein that bound to the RNA genome. (Figure generated using Microsoft PowerPoint).
Fig. 1. The structure of SARS-CoV-2 as a virion, including envelope (E), spike (S), and membrane (M) proteins incorporated into SARS-CoV-2 envelope, and nucleocapsid (N) protein that bound to the RNA genome. (Figure generated using Microsoft PowerPoint).SARS-CoV-2 S glycoprotein forms homotrimers that protrude from envelope and then give rise to the coronal appearance, which can bind to the peptidase domain of human angiotensin-converting enzyme 2 (ACE2) as a host cell receptor on the cell membranes of type 2 pneumocytes, to invade susceptible cells for SARS-CoV-2 viral entry (Fig. 2) [1]. The trimeric complex composed of 1,273 amino acids while structurally belongs to the class I membrane fusion protein, where each protomer is functionally categorized into two distinctive subunits: N-terminal S1 subunit and C-terminal S2 subunit be parted by a furin cleavage site. The S1 region mainly includes the roughly 200-residue receptor-binding domain (RBD) for the interaction with the host cell receptor, while the S2 region holds the membrane fusion machinery, encompassing the hydrophobic fusion peptide and two heptad repeats, HR1 and HR2 that can interact to form six-helical bundle as a post-fusion structure [1,[35], [36], [37]]. Receptor binding by RBD in S1 subunit, proteolytic processing at the furin cleavage site between the S1 and S2 subunits via host cell transmembrane serine protease 2 (TMPRSS2) followed by S1 subunit shedding, S2 subunit structural rearranging into stable post-fusion conformation, the fusion of the viral membrane with the host cellular membrane are regarded as the key events for facilitating subsequent viral entry into the host cell [37], [38], [39].
 Fig. 2. The structure of SARS-CoV-2 spike protein comprises N-terminal S1 subunit (highlighted in orange) and C-terminal S2 subunit (yellow), in which S1 contains RBD (red). The RBD attaches to human ACE2 receptor on type 2 pneumocyte (lung epithelial cell) to initiate viral infection. (Figure generated using Microsoft PowerPoint).
Fig. 2. The structure of SARS-CoV-2 spike protein comprises N-terminal S1 subunit (highlighted in orange) and C-terminal S2 subunit (yellow), in which S1 contains RBD (red). The RBD attaches to human ACE2 receptor on type 2 pneumocyte (lung epithelial cell) to initiate viral infection. (Figure generated using Microsoft PowerPoint).RBD, as a globular domain positioned on the distal surface of SARS-CoV S, MERS-CoV S, and SARS-CoV-2 S, has undergone conformational rearrangements in a dynamic manner by interchangeably masking and presenting their receptor-binding interfaces as well as neutralizing epitopes to either host cells or potential neutralizing antibodies [36]. Initial cryogenic electron microscopy (cryo-EM) characterization of the SARS-CoV-2 spike in the pre-fusion conformation revealed two distinctive configurations adopted by the RBDs: in the ‘up’ state, RBD is away from the spike protein that it can engage ACE2 without steric clash; in the ‘down’ state, RBD is tightly packed against the top of the S2 subunit that it prevents ACE2 binding; with the similar phenomena observed as well in SARS-CoV S and MERS-CoV S [35,40,41]. In a receptor-binding event, the RBD would be trapped in the energetically unstable ‘up’ state, towards the gradual destabilization of S1 until S2 is eventually triggered to initiate membrane fusion [42]. Throughout the life cycle of virus, the spike trimer exists in an equilibrium between an inactive, closed conformation with all RBDs in the ‘down’ states and an active, open conformation with the RBDs in mixed ‘up down’ states. The SARS-CoV-2 S protein predominantly shown an asymmetrical homotrimer, with one RBD in ‘up’ state is ACE2-accessible while the other two RBDs in ‘down’ states are not [35].
SARS-CoV-2 RBD comprises residues 319 to 541, including the receptor-binding motif (RBM) spanning residues 438 to 506 which contains major ACE2-contacting residues: ACE2 interacts with residue F486 protruding from the 481 to 487 loop of SARS-CoV-2 RBD, for instance [37,43,44]. The SARS-CoV-2 variants of concern, B.1.1.7, B.1.351, P.1, B.1.617.2, and B.1.1.529, are known to carry several mutations in RBD: B.1.1.7 with N501Y mutation; B.1.351 with K417N, E484K, and N501Y mutations; P.1 with K417T, E484K, and N501Y mutations; B.1.617.2 with L452R and T478K mutations; B.1.1.529 with G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493K/R, G496S, Q498R, N501Y, and Y505H mutations [45], [46], [47], [48], [49], [50]. Owing to viral components undergo structural changes, SARS-CoV-2 variants retain the same host cell receptor binding, however might no longer be recognized by the specific antibodies [51].
The RBD of SARS-CoV-2 spike protein involves in ACE2 receptor engagement to initiate viral infection. SARS-CoV-2 RBD-based immunogens were shown to stimulate the generation of neutralizing sera in animals, indicates that RBD contains immunodominant epitopes capable to elicit neutralizing antibodies that can provide protection against SARS-CoV-2 infection [52]. Taking these factors into account, RBD is a potential therapeutic target for the development of broadly neutralizing antibodies to stop and prevent SARS-CoV-2 viral infection.
1.3. Differences between SARS-CoV, MERS-CoV and SARS-CoV-2
COVID-19 caused by SARS-CoV-2 marks the third major coronavirus outbreak since last two decades, following severe acute respiratory syndrome (SARS) caused by SARS-CoV in 2002 and Middle East respiratory syndrome (MERS) caused by MERS-CoV in 2012 (Table 1) [30,53]. SARS-CoV, first identified in Guangdong province of China on November 2002, resulted in the SARS epidemic with over 8,000 infections and a ∼10% fatality rate. The outbreak of SARS disease was ended in 2004 [54]. MERS-CoV, emerged in Saudi Arabia on June 2012, resulted in the MERS epidemic with over 2,500 infections and a ∼35% fatality rate as of July 2022 [55]. SARS-CoV-2, first identified in Wuhan, Hubei province of China on December 2019, resulted in COVID-19 pandemic with over 601.19 million infections and over 6.48 million deaths worldwide, with the numbers still rising as of 2 September 2022 [26]. It was observed that SARS-CoV-2 caused higher proportion of asymptomatic and mild symptomatic infections as compared to SARS-CoV and MERS-CoV [56]. With the active viral replication of SARS-CoV-2 in the upper respiratory tract (URT), leads to the viral shedding begins from incubation period then peaked during the time of symptom onset showing mild or no symptoms, in contrast to SARS-CoV and MERS-CoV with the viral shedding begins from the time of symptom onset then peaked in the second week after symptom onset [56,57]. As a result, pre-symptomatic transmission is rare for SARS-CoV and MERS-CoV, but plays roles in SARS-CoV-2 spread [56]. On the other hand, MERS-CoV with inefficient human-to-human transmission as compared to SARS-CoV and SARS-CoV-2, but leads to higher mortality [58].
Table 1. A summary comparison of three highly pathogenic coronaviruses as the disease causative agents: SARS-CoV, MERS-CoV and SARS-CoV-2.
| Feature | SARS-CoV | MERS-CoV | SARS-CoV-2 |
|---|---|---|---|
| Disease | SARS | MERS | COVID-19 |
| First identified | Guangdong, China (2002) | Saudi Arabia (2012) | Wuhan, China (2019) |
| Taxonomy | Betacoronavirus(Sarbecovirus) | Betacoronavirus(Merbecovirus) | Betacoronavirus(Sarbecovirus) |
| Primary host cell receptor | ACE2 | DPP4 | ACE2 |
| Human-to-human transmission | Efficient | Inefficient | Efficient |
| Viral shedding |
Starts from the time of symptom onset. Peaks in the second week after symptom onset. |
Starts from the time of symptom onset. Peaks in the second week after symptom onset. |
Starts from the incubation period. Peaks around the time of symptom onset. |
| Proportion of asymptomatic and mild symptomatic infections | Low | Low | High |
| Pre-symptomatic transmission | Rare | Rare | Plays roles in viral spread |
| Level of spread | Epidemic | Epidemic | Pandemic |
| Mortality | Moderate | High | Low |
ACE2, angiotensin-converting enzyme 2; COVID-19, coronavirus disease 2019; DPP4, dipeptidyl peptidase 4; MERS, Middle East respiratory syndrome; MERS-CoV, Middle East respiratory syndrome coronavirus; SARS, severe acute respiratory syndrome; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV, MERS-CoV, and SARS-CoV-2 are classified into the genus betacoronavirus in the family Coronaviridae: SARS-CoV and SARS-CoV-2 within the subgenus sarbecovirus, MERS-CoV within the subgenus merbecovirus [33]. By analysing the spike protein sequence among SARS-CoV, MERS-CoV and SARS-CoV-2, SARS-CoV-2 showed greater sequence homology with SARS-CoV (76% identity, 87% similarity) than with MERS-CoV (42% identity, 58% similarity) [59]. Therefore, with the sharing of significant sequence identity between SARS-CoV and SARS-CoV-2 suggests the possibility of cross-reactivity. Nevertheless, there is lower sequence homology within spike N-terminal regions, taking account of the dissimilarity in regions including RBD while correlating to the difference in host cell receptors used [60]. The primary functional host cell receptor for SARS-CoV and MERS-CoV are ACE2 and dipeptidyl peptidase 4 (DPP4), respectively [61,62]. SARS-CoV-2, similar to its closest homolog SARS-CoV, utilizes ACE2 to enter host cell as well; however by 10 to 20-fold greater affinity (equilibrium dissociation constant, KD = ∼15 nM) than SARS-CoV [35,63]. E484, F486, Q493, and N501 are important RBD residues contribute to the stronger binding of ACE2 towards SARS-CoV-2 than that of SARS-CoV [43]. Hence, with a higher affinity towards ACE2 resulted from the sequence changes in RBD may supports SARS-CoV-2 in more efficient host cell entry, drives higher transmissibility of SARS-CoV-2 [38]. Besides, it was suggested that there is a difference in the immunogenicity of RBD between SARS-CoV and SARS-CoV-2 [64]. The glycans at residues N165, N234, N343 of spike protein shield SARS-CoV-2 RBD from the antibodies [65]. A more hidden RBD of SARS-CoV-2 than that of SARS-CoV leads to immune evasion as potential viral strategy [66].
2. Single-domain antibody as an attractive neutralizing solution for COVID-19
Antibody, otherwise known as immunoglobulin (Ig), is a large and complex hetero-tetrameric protein made up of two heavy chains and two light chains. In conventional antibody as exemplified by human IgG, the heavy chain is composed of one variable domain (VH) and three constant domains (CH1, CH2 and CH3), whereas the light chain is composed of one variable domain (VL) and one constant domain (CL) (Fig. 3) [67]. Antibody, produced by the immune system in response to antigens, contributes to confer immunity in the body. For instance during viral infection, the antibodies neutralize viral targets by precisely block the virus-host cell receptor association site, sterically inhibit the host cell receptor binding, or prevent viral entry into the host cell through a variety of molecular mechanisms [36,68]. Hence, neutralizing antibodies potentially protect individuals against viral infection, or block viral entry during the early infection step to suppress virus replication.
 Fig. 3. The structure of conventional antibody and heavy-chain only antibody, which can be split up into antigen-binding fragment (Fab) and crystallisable fragment (Fc). Conventional antibody, as exemplified by human IgG, is composed of two heavy chains and two light chains: each heavy chain is composed of one variable domain (VH) and three constant domains (CH1, CH2 and CH3), whereas each light chain is composed of one variable domain (VL) and one constant domain (CL). Heavy-chain only antibody from camelid is composed of only two heavy chains: each heavy chain is composed of single variable domain (VHH) and two constant domains (CH2 and CH3). Heavy-chain only antibody from cartilaginous fish, termed immunoglobulin new antigen receptor (IgNAR), is composed of only two heavy chains: each heavy chain is composed of single variable domain (VNAR) and five constant domains (CNAR1, CNAR2, CNAR3, CNAR4 and CNAR5). (Figure generated using Microsoft PowerPoint).
Fig. 3. The structure of conventional antibody and heavy-chain only antibody, which can be split up into antigen-binding fragment (Fab) and crystallisable fragment (Fc). Conventional antibody, as exemplified by human IgG, is composed of two heavy chains and two light chains: each heavy chain is composed of one variable domain (VH) and three constant domains (CH1, CH2 and CH3), whereas each light chain is composed of one variable domain (VL) and one constant domain (CL). Heavy-chain only antibody from camelid is composed of only two heavy chains: each heavy chain is composed of single variable domain (VHH) and two constant domains (CH2 and CH3). Heavy-chain only antibody from cartilaginous fish, termed immunoglobulin new antigen receptor (IgNAR), is composed of only two heavy chains: each heavy chain is composed of single variable domain (VNAR) and five constant domains (CNAR1, CNAR2, CNAR3, CNAR4 and CNAR5). (Figure generated using Microsoft PowerPoint).With the cumulative SARS-CoV-2 infections while lacking effective treatments, the transfusion of convalescent plasma containing the existing neutralizing polyclonal antibodies serves as powerful therapeutics for COVID-19 [68]. Polyclonal antibodies, also known as the heterogeneous antibody mixtures, can recognize complex antigens carrying numerous epitopes [69]. It was shown that convalescent plasma inhibited SARS-CoV-2 infection, as well as relieved symptoms in newly infected patients [70,71]. Several studies have demonstrated that the neutralizing antibodies from convalescent plasma of COVID-19 patients potently neutralized SARS-CoV-2 pseudovirus with half-maximal inhibitory concentration (IC50) ranged from 1 to 300 ng/mL, and replication-competent SARS-CoV-2 with IC50 from 15 to 500 ng/mL [72], [73], [74], [75], [76]. However, low titres of neutralizing antibodies in convalescent plasma can poses a risk factor for an antibody-dependent enhancement (ADE) response of viral infection. The desired high-titre convalescent plasma is limited in supply, particularly in the situation with tenuous supply of convalescent plasma for the large amount size of infected patients. Also, there exist challenges for the storage and deployment of convalescent plasma.
The use of laboratory-synthesized neutralizing antibodies helps to avoid infection risks that might occur when using human blood plasma or serum, in addition the antibodies can be applied in smaller quantity. Monoclonal antibody (mAb) is a homogeneous antibody derived from single B lymphocyte clone, with specificity against single epitope on antigens [77]. The genes of mAbs can be cloned from B cells of recovered COVID-19 patients with mounted natural immune response against SARS-CoV-2 [72,73]. The manufacturing of mAbs is independent of donors, moreover the quality of mAbs is easier to control. The development of therapeutic mAbs serves as the frontline in combating COVID-19 [78]. For instance, neutralizing mAb regdanvimab developed by Celltrion Inc. has been approved in South Korea to treat mild COVID-19 in patients aged above 50 years old with underlying health problem such as cardiovascular disease, diabetes or obesity, due to its potency to reduce the risk of progression to severe disease in COVID-19 patients [79,80].
However, mAbs as large biomolecules are intravenously delivered with low efficiency across the plasma-lung barrier for pulmonary infection treatment, thus high administration doses of mAbs in several grams would be required for efficient neutralization [81], [82], [83]. The production of mAbs in large scale usually takes at the minimum of 3 to 6 months, thus would be difficult to achieve a timely production during pandemic [64]. As mAbs undergo post-translational modifications, the production of mAbs require eukaryotic expression system by using mammalian cells that are expensive to maintain [84]. In addition, mAbs can be degenerated due to the exposure to extreme ambient conditions such as humidity and high temperatures [67]. As there are diverse environmental conditions for varying countries, while there is the requirement for specific temperature to be maintained during storage and transportation of mAbs, widespread clinical use of mAbs may be limited. With the aim to improve properties in therapeutic applications, recombinant antibodies are generated using molecular lab techniques. Antibody fragments or domains, such as Fab (antigen-binding fragment, 50 kilodaltons (kDa)), scFv (single-chain variable fragment, 30 kDa), and VH (heavy chain variable domain, 15 kDa) are appealing antibody formats to be used as smaller biomolecules for therapeutics [85].
In the early 1990s, camelids or cartilaginous fish were found to be possessing unconventional antibodies in their immune system: exists as a homodimer that is naturally devoid of light chains, thus known as heavy-chain only antibodies [86]. A single variable domain (VHH from camelid or VNAR from cartilaginous fish) represents the antigen-binding region of heavy-chain only antibodies, in lieu of two variable domains (VH and VL) that typically forms the antigen-binding region of conventional IgG (Fig. 3). VHH contains three hypervariable loops, denoted complementarity-determining region 1 (CDR1), CDR2 and CDR3; VNAR contains four hypervariable loops, denoted CDR1, hypervariable region 2 (HV2), HV4 and CDR3 [32,67]. CDR3 comprises the most variable region in antibody, also with at least 60 to 80% of the contact with the antigen, thus mainly contributes to the specific binding of antibodies towards target antigens [20,87,88]. The single variable domain from heavy-chain only antibodies can be expressed independently as a ∼12 to 15 kDa antibody fragment, with the acquired specificity and affinity for target antigen is comparable to the conventional antibodies, therefore contain autonomous function as single-domain antibodies (sdAbs) (Fig. 4) [86,89,90].
 Fig. 4. Representation of single-domain antibody (sdAb), as exemplified by the single variable domain (VHH) from camelid heavy-chain only antibody, which exhibit autonomous function as an antibody: with CDR1, CDR2 and a long protruding CDR3 (highlighted in purple, green and red, respectively) to control the antigen binding. The blocking of ACE2-RBD interaction by sdAb serves as one of the potential neutralization mechanism by sdAbs against SARS-CoV-2. (Figure generated using Microsoft PowerPoint).
Fig. 4. Representation of single-domain antibody (sdAb), as exemplified by the single variable domain (VHH) from camelid heavy-chain only antibody, which exhibit autonomous function as an antibody: with CDR1, CDR2 and a long protruding CDR3 (highlighted in purple, green and red, respectively) to control the antigen binding. The blocking of ACE2-RBD interaction by sdAb serves as one of the potential neutralization mechanism by sdAbs against SARS-CoV-2. (Figure generated using Microsoft PowerPoint).Single-domain antibody, with a molecular weight of ∼12 to 15 kDa, is approximately one-tenth of the size of a conventional IgG in ∼150 kDa [90,91]. As small biomolecules, sdAbs exhibit efficient tissue penetration [92,93]. sdAbs are less affected by steric hindrances that interfere with the binding for large conventional antibodies, as a result sdAbs have larger number of accessible epitopes [94,95]. In addition, with an extended antigen-binding region due to a long protruding CDR3 loop, sdAbs are capable to access cryptic epitopes [96,97]. Hence, sdAbs retain full antigen-binding capacity of antibody. Small size sdAbs allow for rapid kilogram-scale production with ease in prokaryotic expression systems, leads to high yield with relatively low production cost, consequently enable fast implementation during the outbreak [94,98]. As non-complex structure, sdAbs can be expressed in yeast and mammalian cells as well [99], [100], [101]. Since sdAbs do not bind light chains, with the absence of hydrophobic interface between VH and VL domain render the sdAbs a more hydrophilic surface, thus have high solubility for ease of downstream processing [102], [103], [104]. The intrinsic stability of sdAbs as exemplified by the inherent thermostability and chemostability, enables sdAbs to withstand prolonged storage [36,68,105,106]. For instance, sdAbs are with tolerance towards pH ranging from 3 to 11, also with resistance to chemical denaturant (0.35 - 8 M urea) [[107], [108], [109],105,110]. Therefore, sdAbs can be reserved as a stockpile of therapeutic options for future epidemic.
In addition, the small size and non-complex structure of sdAbs allow flexible formatting according to the needs [68,98]. The origin of sdAbs from animals may limit their therapeutic application in humans, as there is immunogenicity risk. Thus, humanization techniques have been adopted, by modifying the animal-specific amino acid sequences within framework into the human heavy chain variable domain as its counterpart, to reduce species heterogeneity without altering its antigen-binding affinity and solubility [94,111,112]. The monomeric nature of sdAbs has its drawback, such as their binding kinetics in terms of fast dissociation rates (koff) may reduce neutralization potency. Therefore, sdAbs can be multimerized to enhance avidity, such that sdAbs that are designed in homo-dimeric or homo-trimeric form can increase valency to improve antiviral activities, while sdAbs that are designed in heterodimeric form can simultaneously targeting different epitopes to prevent virus mutational escape [113]. sdAbs have short serum half-life and rapid renal clearance due to their small size, as limitations for treatment and prevention of viral disease [67,114]. Hence, sdAbs can be fused with the crystallisable fragment (Fc) of IgG to become larger protein, to extend their blood residential time as well as prolong their circulation in the body [93,115].
In 2019, FDA has given approval to the first sdAb-based medicine, caplacizumab for the treatment of acquired thrombotic thrombocytopenic purpura, with an estimated cost of $270,000 [116,117]. The innovative nature and drug development account for the high cost of caplacizumab, nonetheless the novel therapy represents a major breakthrough [117]. Besides being used as injectable drug, the small and stable sdAbs may be nebulized and administered via inhalation directly to the airway epithelia, which can maximize their bioavailability and function by having high concentration of therapeutics at the respiratory site infection [64,118]. It was reported that an inhaled sdAb, ALX-0171 for the treatment of respiratory syncytial virus has entered clinical trials [119]. Therefore, the use of sdAbs as biologics is an interesting approach, particularly for the treatment of respiratory infection. Similarly, with the generation of a neutralizing inhaler containing sdAbs offers a possibility for directly blocking viral replication in the upper airway during the early stages of COVID-19, meanwhile it helps to improve patient compliance by being a needle-free treatment.
2.1. Case studies on broadly-neutralizing single-domain antibody for SARS-CoV-2
Surface display technology has been utilized for the selection of sdAbs specific for the targeted antigen. There are several antibody surface display technologies, included phage display, ribosome display, yeast surface display, and bacterial surface display [13,[120], [121], [122]]. As exemplified by the widely employed phage display technology (Fig. 5), the gene encoding for sdAb is fused with the gene encoding for bacteriophage's coat protein, giving rise to the display of sdAb on bacteriophage's surface in which can be applied for the selection of antigen-specific binders [123]. Afterwards, a library of sdAbs genes are cloned into phagemid vectors, lead to the generation of a sdAb library with diversity. There are multiple types of sdAb library, inclusive of immune library and non-immune library such as naïve or synthetic library. sdAb library has the potential to be a rapidly accessed resource, which may bring about the fast-track discovery of neutralizing antibodies during an outbreak.
 Fig. 5. Overview of the process from sdAb generation to phage-displayed immune sdAb library construction. A camelid was immunized for few times (within ∼35 days) with the inactivated SARS-CoV-2 or RBD as antigen, to produce specific antibodies against SARS-CoV-2. After the last immunization, blood was obtained from immunized camelid, with peripheral blood lymphocytes containing antibody gene were isolated, while total RNAs were extracted to be used as template for synthesizing complementary deoxyribonucleic acid (cDNA). The VHH or sdAb coding regions were then cloned into phagemid vectors. Various sdAb coding regions were amplified by polymerase chain reaction (PCR) while undergo cloning, for the construction of a recombinant DNA library to be expressed via phage display (library size ∼1010), with each phage expresses sdAb copies on its surface. (Figure generated using Microsoft PowerPoint).
Fig. 5. Overview of the process from sdAb generation to phage-displayed immune sdAb library construction. A camelid was immunized for few times (within ∼35 days) with the inactivated SARS-CoV-2 or RBD as antigen, to produce specific antibodies against SARS-CoV-2. After the last immunization, blood was obtained from immunized camelid, with peripheral blood lymphocytes containing antibody gene were isolated, while total RNAs were extracted to be used as template for synthesizing complementary deoxyribonucleic acid (cDNA). The VHH or sdAb coding regions were then cloned into phagemid vectors. Various sdAb coding regions were amplified by polymerase chain reaction (PCR) while undergo cloning, for the construction of a recombinant DNA library to be expressed via phage display (library size ∼1010), with each phage expresses sdAb copies on its surface. (Figure generated using Microsoft PowerPoint).2.2. Camelid VHH against SARS-CoV-2 and other coronaviruses
According to the work by Wrapp et al. [36], SARS VHH-72, with high affinity to SARS-CoV RBD (KD = 1.2 nM), was identified via phage-displayed sdAb library derived from a llama immunized with SARS-CoV and MERS-CoV S protein. Camelid VHH domains have high degree of homology with human type 3 VH domains, thus with the high conservation leads to low immunogenicity [124]. Crystal structure of SARS VHH-72 bound to viral target revealed that the epitope of SARS VHH-72 did not overlap with the ACE2 binding site on the SARS-CoV RBD. Instead, there were steric clashes between ACE2 and SARS VHH-72, possibly cause interferences towards ACE2 binding to RBD. SARS VHH-72 has shown the ability to cross-react with the SARS-CoV-2 RBD (KD = 39 nM), as well as could interfere with the ACE2 binding. It is postulated that SARS VHH-72 binds onto the part of SARS-CoV RBD sharing low sequence variation with SARS-CoV-2 RBD, thus able to broadly bind towards SARS-CoV-like viruses.
Further engineering of the cross-reactive SARS VHH-72 into a bivalent and monomeric human IgG Fc-fusion has conferred it the ability to readily neutralize SARS-CoV-2 pseudovirus, with an IC50 of approximately 200 ng/mL. SARS VHH-72-Fc, through the fusion to human IgG1 Fc domain, can interact with Fc receptor (FcR) expressed on immune cells such as macrophages, B cells and monocytes. The engagement of FcR activates the immune cells to get rid of viruses inside the body, with Fc-dependent cytotoxic functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and antibody-dependent cellular phagocytosis (ADCP) [125], [126], [127]. The multi-valency approach aimed to enhance avidity, and to circumvent ADE of viral infection that can be triggered instead through Fc-FcR interaction due to the sub-optimal antigen–antibody interactions [128]. The production of SARS VHH-72-Fc has reached expression levels of ∼300 mg/L in the industry's standard Chinese hamster ovary (CHO) cell system, which is high-yielding. The SARS-CoV-2 variants of concern, B.1.1.7, B.1.351, P.1, B.1.617.2, and B.1.1.529 share one specific mutation within the spike protein, called D614G [49,129]. D614G mutant exhibits greater trimeric spike protein stability, results from the greater incorporation of spike protein into virions with less S1 shedding [130]. D614G mutant increases the viral infectivity without any effect on pathogenesis [131], [132], [133]. Nieto et al. [122] reported that the monomeric Fc fusion of SARS VHH-72 exhibiting modest neutralization for authentic SARS-CoV-2 wild type (D614) and D614G mutant (G614), with IC50of 1,287.75 nM and 1,233.90 nM, respectively. The hexavalent Fc fusion of SARS VHH-72 engineered by Zupancic et al. [134] potently neutralized SARS-CoV-2 pseudovirus variants B.1.1.7 and B.1.351, with IC50 of 310 pM and 72 pM, respectively.
Xiang et al. [113] discovered that the serum of a llama immunized with SARS-CoV-2 RBD contained potent neutralizing sdAbs with picomolar to femtomolar affinities towards the RBD, such as Nb20 (KD = 10.4 pM) and Nb21 (KD < 1 pM) neutralized SARS-CoV-2 pseudovirus with IC50 of 102 pM and 45 pM, respectively. Structural characterization through cryo-EM showed that Nb20 or Nb21 binds to epitope that partially overlapping with the ACE2 binding site on RBD, with their CDR1 and CDR3 would clash with the ACE2 α-1 helix containing major portion of residues responsible for the coordination of ACE2-RBD interaction, which can cause steric interference towards ACE2 binding when bound to the RBDs in ‘up’ states. Meanwhile, Nb20 or Nb21 can bind to the RBDs in ‘down’ states as well, indicates with the concurrent binding of three Nbs to all three RBDs in ‘down’ states may be locking the spike into an inactive conformation. The homo-trimeric constructs based on Nb20 and Nb21 have shown up to ∼30-fold improvement of neutralization potency towards SARS-CoV-2 pseudovirus as compared to that of their monomeric form, with IC50 of 4.14 pM and 1.32 pM, respectively. Besides, Nb20 and Nb21 potently neutralized SARS-CoV-2 Munich strain (D614G mutant) with IC50 of 48 pM and 22 pM, respectively, while the homo-trimeric constructs of Nb20 and Nb21 have shown up to a ∼6-fold increase of neutralization potency towards SARS-CoV-2 Munich strain with IC50 of 5.43 pM and 6.04 pM, respectively [113,135]. Nb20 and Nb21 in both monomeric and homo-trimeric form presented thermal stability ranged from 70 to 72.8 °C. Xiang et al. [113] also identified Nb34 and Nb95 which can only bind with at least two RBDs in ‘up’ states, with epitopes that do not overlap with the ACE2 binding site. Nb34 is fitted onto the top of spike trimer, locking the helices of S2 at pre-fusion conformation and thus prevent membrane fusion; Nb95 is accommodated nearby to the firmly fixed N-terminal domain (NTD) of spike trimer, which may restrict spike flexibility. It was demonstrated that Nb34 and Nb95 can neutralize SARS-CoV-2 Munich strain as well, with IC50of 1.125 nM and 5.105 nM, respectively.
The selection of sdAbs from a synthetic library requiring at most 2 to 3 weeks, as compared to the traditional generation of sdAbs with at least 6 weeks for animal immunization followed by within 3 to 4 months for antibody selection [136]. Schoof et al. [121] utilized a yeast surface-displayed synthetic llama sdAb library, where an anti-SARS-CoV-2 RBD Nb6 (KD = 41 nM) that inhibited SARS-CoV-2 pseudovirus infection with IC50 of 2,000 nM was identified. Cryo-EM showed that Nb6 binds to spike in completely inactive conformation by recognizing RBD epitope that overlap with ACE2 binding site. It was observed that one Nb6 takes a straddle position at the interface between two ‘down’-state RBDs, with its CDR3 reach over to the neighbouring RBD: indicates neutralization mechanism by locking the two RBDs into ‘down’ states while can pre-organize the binding site for a second and third Nb6 molecule, hence will stabilize the closed spike conformation that renders RBDs inaccessible to ACE2 binding. With affinity maturation of Nb6 by mutagenesis of CDR1 and CDR3 region, in addition with multivalency design, resulted in generating a matured Nb6 in trivalent form (mNb6-tri). mNb6-tri neutralized SARS-CoV-2 pseudovirus with IC50 of 120 pM, showing up to ∼104-fold enhanced neutralization potency compared to that of Nb6. It is predicted that the neutralization mechanism of mNb6-tri involves conformational control of RBD accessibility, in which one mNb6-tri can simultaneously lock all three RBDs into ACE2-inaccessible ‘down’ states. Furthermore, the monovalent Nb6 and engineered mNb6-tri neutralized authentic SARS-CoV-2 (isolate France/IDF0372/2020, V367F mutant) with IC50 of 3,300 nM and 54 pM, respectively [121,137]. mNb6-tri retains function after the heat treatment for an hour at 50 °C, aerosolization, as well as lyophilization.
Gai et al. [20] have isolated Nb11-59 specific for SARS-CoV-2 RBD (KD = 21.6 nM) while exhibited the most potent neutralizing activity against authentic SARS-CoV-2 with 50% neutralizing dose (ND50) of 550 ng/mL, via phage-displayed sdAb libraries generated from camels immunized with SARS-CoV-2 RBD. Nb11-59 was shown to block the interaction between human ACE2 with the RBD of closely related beta-coronaviruses: bat-SL-CoV-WIV1 RBD and SARS-CoV RBD; as well as can block the interaction between ACE2 and eight SARS-CoV-2 RBD mutants, including Q321L, V341I, N354D, V367F, K378R, V483A, Y508H, and H519P circulated in China, England, France, and the United States. [138,139]. Humanized Nb11-59 (HuNb11-59) can be mass-produced using the methylotrophic yeast Pichia pastoris, with 99.36% purity and 20 g/L yield. High drug stability of HuNb11-59 has been proven, with a good stability profile at temperature ranged from 4 to 40 °C, also with a consistent post-nebulization stability profile showing merely small aggregates (0.23%) formed after nebulization.
Nieto et al. [122] developed single-step sdAb selection using Escherichia colisurface-displayed sdAb library derived from an alpaca immunized with SARS-CoV-2 S protein, coupled with non-complex density gradient centrifugation. The bacterial surface-displayed system utilizes the high transformation efficiency of Escherichia coli, neither the infection by bacteriophages nor the shuttling into yeast cells is required for the surface display of sdAbs [140]. W25, which targets SARS-CoV-2 RBD (KD = 295 pM), efficiently competed against ACE2 for binding to RBD with an EC50 of 33 nM. W25 neutralized authentic SARS-CoV-2 wild type and D614G mutant, with IC50 of 9.28 nM and 5.09 nM, respectively. W25Fc, as a dimeric Fc fusion of W25, had a better neutralizing performance for authentic SARS-CoV-2 wild type and D614G mutant with IC50of 7.39 nM and 3.69 nM, respectively. Interestingly, there was a slight enhancement in neutralization effect towards the D614G mutant. In addition, the effective conjugation by covalently labelled W25 to Horseradish Peroxidase (HRP) may be useful for the diagnostic development involving direct antigen detection.
Pymm et al. [44] have identified the four most potent sdAbs against SARS-CoV-2 RBD using the phage-displayed sdAb libraries generated from alpacas immunized with spike protein from SARS-CoV-2 and RBD from SARS-CoV and SARS-CoV-2: WNb 2 (KD = 360 pM), WNb 7 (KD = 260 pM), WNb 15 (KD = 140 pM), and WNb 36 (KD = 430 pM). The sdAbs bound to RBD can be divided into two major groups: Cluster 1 sdAbs, as exemplified by WNb 2 and WNb 36 did not compete with Cluster 2 sdAbs, as exemplified by WNb 7 and WNb 15 for RBD binding. Structural characterization of sdAbs-RBD complex revealed that Cluster 1 sdAb and Cluster 2 sdAb bound simultaneously to two distinct antigenic sites on RBD, with each epitope overlapped with the ACE2 binding region on RBD at different degrees. Cluster 1 sdAb, with the epitope overlapping the binding position of ACE2 α-1 helix as the primary binding site for RBD, possibly contribute to ACE2 blocking. Cluster 2 sdAb, with the epitope overlapping the position of ACE2 α-10 helix considered as small binding overlap on the RBD, but can bind to RBD in an orientation that will cause steric clashes towards ACE2-RBD binding. Remarkably, the dimeric Nb-Fc fusions, WNbFc 7 and WNbFc 15 inhibited ACE2 interaction with SARS-CoV S1 at IC50 of 830 pM and 1.45 nM, respectively. N501Y mutation was found in the SARS-CoV-2 variants B.1.1.7, B.1.351, P.1 and B.1.1.529 [49,[141], [142], [143]]. N501, as one of the six key ACE2-contacting residues inside the RBD, is important for ACE2-RBD interaction [144,145]. With the N501Y mutation, results in the higher binding affinity of RBD for ACE2, which leads to increasing viral transmissibility[146,147]. The two-antibody mixture combination, WNbFc 36 + 7 and WNbFc 2 + 15 neutralized authentic SARS-CoV-2 D614G N501Y mutant as well as the wild type SARS-CoV-2, with IC50 of ∼100 pM and ∼300 pM, respectively. Prophylactic administration of WNbFc 36 + 7 at a dose of 0.2 mg/kg has decreased the viral RNA load in the lung by up to 104-fold in the SARS-CoV-2 D614G N501Y mutant-infected mice at 3 days’ post infection (dpi), thus displayed the potential of antibody cocktails as prophylactic agents against SARS-CoV-2 in vivo.
Koenig et al. [148] have selected VHH E (KD = 2 nM) and VHH V (KD = 9 nM) as high affinity binders specific for SARS-CoV-2 RBD via phage-displayed sdAb libraries generated from an alpaca and a llama immunized with SARS-CoV-2 RBD and inactivated SARS-CoV-2. VHH E and VHH V neutralized SARS-CoV-2 pseudovirus with IC50 at 60 nM and 198 nM, respectively, likewise neutralized authentic SARS-CoV-2 wild type with IC50 at 48 nM and 142 nM, respectively. X-ray crystallography revealed two distinctive binding epitopes on the RBD: VHH E binds to the ACE2 binding site on RBD, possibly block ACE2 binding; VHH V binds to RBD will cause steric clash with the ACE2 glycans at N322 and N546, possibly interfere with ACE2 binding. Cryo-EM revealed that the binding of VHH E trapped the RBDs in the ‘up’ states, leads to the stabilization of spike in a conformation with all three RBDs in ‘up’ states, as well as triggering activation of the fusion machinery in spike without host cell contact by ACE2 receptor, resulted in the spike undergo a premature transition from pre-fusion conformation into the non-reversible post-fusion conformation. The non-productive fusion caused the virions to be non-infectious. Engineered biparatopic VHH VE neutralized SARS-CoV-2 pseudovirus and authentic SARS-CoV-2 wild type with IC50 of 4.1 nM and 1.32 nM, respectively, displayed up to 50-fold improved neutralization potency compared to its monovalent form. The simultaneous targeting on two different epitopes by VHH VE, in addition with the aberrant activation of the spike fusion machinery as the mechanism of neutralization, suppressed the emergence of escape mutants during experimental evolution.
Xu et al. [149] demonstrated that mice can be engineered to produce camelid VHHs, known as nanomice. The anti-SARS-CoV-2 RBD nanobodies, Nb12 (KD = 30 nM), Nb15 (KD = 8.15 nM), Nb30 (KD = 6.55 nM) and Nb56 (KD = 3.26 nM) were discovered via phage-displayed sdAb libraries generated from nanomice and llama immunized with SARS-CoV-2 RBD and S protein, shown to neutralize SARS-CoV-2 pseudovirus with IC50 values ranging from 320 pM to 7.145 nM. Structural characterization of sdAbs-RBD complex revealed that Nb15 and Nb56 recognize the RBD-ACE2 interface, possibly neutralize by blocking ACE2 binding; Nb12 and Nb30 recognize a conserved region on RBD without overlapping with the ACE2 binding site, neutralize by sterically interfere with ACE2 binding. The trivalent Fc fusion of Nb12, Nb15, Nb56 and bivalent Fc fusion of Nb30 neutralized SARS-CoV-2 pseudovirus with IC50 values ranging from 43 to 614 pM, as well as neutralized authentic SARS-CoV-2 wild type and variants B.1.1.7, B.1.351, P.1 with IC50 values ranging from to 3 pM to 9.374 nM. Moreover, trivalent Nb12-Fc and bivalent Nb30-Fc neutralized SARS-CoV, Bat-SL-CoV (WIV1, WIV16, SHC014, LYRa11, Rs7327, Rs4084), Bat-CoV-RaTG13, Pangolin-CoV-GD and Pangolin-CoV-GX pseudoviruses at IC50 values below 423 pM. Based on informatics analysis, the binding epitopes for Nb12 and Nb30 are 54% and 79% conserved among sarbecoviruses, respectively, in comparison to the 23% conserved binding epitope for 51 RBD-directed human antibodies on average. The nanobodies have shown good stability profile, with their neutralization activity retained after nebulization, also with their integrity maintained after the heat treatment for 10 min at 98 °C.
2.3. Shark VNAR against SARS-CoV-2 and other coronaviruses
The first study on neutralizing sdAbs from shark origin, VNAR against SARS-CoV-2 was presented by Gauhar et al. [150]. Through the screening of phage-displayed semi-synthetic shark VNAR libraries, followed by further reformatting of the isolated VNARs into bivalent human IgG Fc-fusion, VNAR-hFc antibodies specific to SARS-CoV-2 RBD were obtained: 3ID10_16, 6ID10_75 and 3ID10_99. These antibodies blocked the interaction between ACE2 and wild type RBD at IC50 values ranging from 2.5 to 130 nM, with neutralizing potential towards the authentic SARS-CoV-2 wild type. The SARS-CoV-2 variants B.1.351 and P.1 harbouring the E484K mutation, which connected to the immune escape from neutralizing antibodies induced by prior infection and SARS-CoV-2 reinfection [151], [152], [153]. Evidently, 3ID10_16, 6ID10_75 and 3ID10_99 possessed the blocking ability for two RBD mutants including E484K and N501Y. The discovery of shark VNAR as a novel class of sdAbs against SARS-CoV-2 has expanded the molecular toolkit of potential therapeutics for COVID-19.
Ubah et al. [154] described the identification of monomeric VNARs: 3B4 (KD = 17.2 nM) and 2C02 (KD = 63 nM) from phage-displayed synthetic shark VNAR library screened against SARS-CoV-2 RBD, to be discovered as the potent neutralizers of authentic SARS-CoV-2 wild type at IC50 of 11.5 nM and 839 pM, respectively. The VNARs also effectively neutralized WIV1-CoV and SARS-CoV pseudoviruses, with IC50 ranging from 7.93 to 71.1 nM. Of significance, 3B4 was capable of neutralizing MERS-CoV pseudovirus at IC50 of 1,050 nM, suggesting that 3B4 bound to conserved region among beta-coronaviruses. Crystallographic analysis of 3B4 and 2C02 revealed that each VNARs recognizing distinctive epitopes on the RBD, neither of which overlaps with RBD-ACE2 interface. 3B4 binds distal to the ACE2 binding site on the ‘up’-state RBD, neutralizes by resulting in steric clash with ACE2 directly. It was observed that 2C02 can bind to the RBDs in either ‘up’ or ‘down’ state: 2C02 binds to the ‘up’-state RBD without close contact to ACE2, possibly neutralizes by causing allosteric effects towards ACE2 binding; 2C02 also binds within a cleft formed between protomer 1’s ‘down’-state RBD and protomer 3’s NTD, neutralizes by securing RBD in the ‘down’ state to block the access for ACE2. The study features shark VNARs as the useful therapeutic agents for beta-coronaviruses.
According to the work by Feng et al. [155], 20G6 and 17F6 were isolated via phage-displayed shark VNAR library derived from a bamboo shark immunized with SARS-CoV-2 S protein. Structural characterization of VNAR-RBD complex revealed that 20G6 and 17F6 contain “WXGY” motif within the CDR3, that can bind to the residues 365 to 380 of RBD in ‘up’ state without overlapping with the ACE2 binding site, neutralize by causing steric hindrance towards ACE2-RBD interaction. Furthermore, the binding epitopes for 20G6 and 17F6 are highly conserved among sarbecoviruses. The dimeric Fc fusion of 20G6 and 17F6, 20G6-Fc (KD < 10 pM) and 17F6-Fc (KD < 10 pM) neutralized Pangolin-CoV-GD1 and Bat-CoV-RaTG13 pseudovirus, as well as SARS-CoV-2 pseudovirus wild type and variants B.1.351, B.1.617.2, B.1.617.2.1, B.1.617.1, C.37 at IC50 values below 10 nM. Besides, authentic SARS-CoV-2 wild type and variants B.1.351, B.1.617.2 can be neutralized by 20G6-Fc at IC50 ranging from 9.36 to 11.79 nM, as well as neutralized by 17F6-Fc at IC50 ranging from 19.87 to 34.36 nM. Intranasal delivery of 20G6-Fc at 10 mg/kg conferred protection prophylactically and therapeutically, by reducing viral RNA load and lung pathology without significant weight loss in SARS-CoV-2-infected mice and SARS-CoV-2 variant B.1.351-infected mice at 3 dpi. High thermal stability of the VNARs has been proven, with strong binding activity retained after the heat treatment for an hour at 90 °C.
Table 2. A summary on the characteristics of previously reported sdAbs against SARS-CoV-2, including binding affinity towards RBD (KD) and neutralization potency are presented.