1. Introduction
Despite decades of industry’s efforts, corrosion-related issues continue to be recognized as one of the foremost issues in LWRs. For example, at the international symposium AREVA-Germany presented a worldwide status overview in which 14,000 EIS (Events of Interest for Safety) were identified in the IAEA’s nuclear events database as of 2008 (Fig. 1) (ISaG-2013). Due to the potential degradation of reactor pressure boundaries, it is now mandatory to schedule an in-service inspection to monitor the development of unexpected degradation.
 Fig. 1. Corrosion –major cause of degradation of LWRs worldwide. (Reproduced from the distribution at ISaG-2013: “Analysis and assessment of the risk potentials caused by corrosion in German LWR plants”). Flow accelerated corrosion (FAC) is the major cause but stress corrosion cracking (SCC) induces large safety impacts due to its potential failure before leak.
Fig. 1. Corrosion –major cause of degradation of LWRs worldwide. (Reproduced from the distribution at ISaG-2013: “Analysis and assessment of the risk potentials caused by corrosion in German LWR plants”). Flow accelerated corrosion (FAC) is the major cause but stress corrosion cracking (SCC) induces large safety impacts due to its potential failure before leak.2. Basic corrosion theories for LWRs1
Corrosion of nuclear materials, i.e. the interaction between these materials and their environments used in LWRs, is a major issue for plant safety as well as for operation and economic competitiveness. With this acknowledgement, Damien Féron recently performed a critical review of the fundamental corrosion mechanisms that affect nuclear power plants and facilities. This was complemented by reviews of monitoring and control methodologies, as well as modeling and lifetime prediction approaches (Féron, 2012). However, the effect of radiation on corrosion is not designated as the root cause of many of these corrosion issues. Although the radiation effect is hidden behind electrochemical corrosion potential (ECP) induced by radiation, his review does not explain the accelerated corrosion mechanism due to the formation of the closed circuit with a “differential radiation cell (DRC)” which is one of the main topics of this report. This concept is an adaptation of the “differential aeration cell” which is widely applied in conventional corrosion studies (NACE, 1959). However this mode of corrosion has been dismissed in the nuclear community considering that the transport of ions with flow is unlikely due to the high purity of reactor water which is different from an electrolyte. The author found that the analogous mechanism should be occurring in reactor water with radiation differences and should be inducing severe interaction between the structural materials and LWR’s through de-passivation followed by “radiation-induced electrolytic (RIE)” corrosion.
2.1. “Radiation-induced electrolytic corrosion” hypothesis
The “Long-Cell Action (LGCA) Corrosion” as summarized in Annex A is well established as the basic theory behind cathodic protection with a more easily corroded “sacrificial metal“ that acts as the anode in soil corrosion. This theory was originally developed by Pope (1956) followed by Schaschl and Marsh (1963), based on 45 years of research by the U.S. National Bureau of Standards (NBS, currently NIST) (NACE, 1959). The NBS was using the terminology “differential aeration cell” which causes the corrosion of metals as a result of the formation of an oxygen concentration cell induced by an uneven supply of air on the metal surface exposed to wet soils. However, NBS’ “differential aeration cell” concept did not include the LGCA mechanism proposed by R. Pope. Beginning with these works, the author has been investigating the potential involvement of the “differential radiation cell (DRC)” which is an extension of LGCA to the reactor environment.
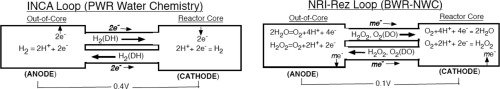
This mode of corrosion cell has been dismissed in the nuclear community considering that the transport of ions with flow is unlikely due to high purity of reactor water. The author recently found that the corresponding mechanism is occurring in reactor water with a radiation difference. This phenomenon should induce severe interaction between structural materials and the environments employed in LWRs through “radiation-induced electrolytic (RIE)” corrosion mechanism. The contemplated mechanism is illustrated in Fig. 2 where the reactor core region as well as the out-of-core region are simplified with a point model.

Fig. 2. Radiation-induced electrolytic (RIE) phenomena.
INCA loop (PWR water chemistry)
-
(1)
The top figure illustrates the RIE phenomenon observed in the INCA loop which was tested by simulating the PWR reactor water chemistry.
-
(2)
The in-core redox water potential was observed to be approximately + 0.3V higher than the out-of-core region. The high redox water potential attracts conduction electrons through piping into the redox water, reducing H+ ions into dissolved hydrogen (DH).
-
(3)
The outflow from the reactor region carries the valence electrons with an neutral DH to the out-of-core region where the dissolved hydrogen is oxidized to hydrogen ions at the metallic surface of the loop, releasing electrons which are transported back to the reactor region as conduction electrons. The out flow from the out-of-core region carries hydrogen ions. Thus both the electric charge balance as well as the material balance are established comprising of a closed electric circuit.
NRI-Rez loop (BWR-NWC)
-
(4)
The bottom figure illustrates the RIE phenomenon observed in the NRI-Rez loop which was tested by simulating the BWR-NWC condition. In this case the cathodic reactions in the core region involve two reactions with O2 and H2O2. The counter cell reactions also involve two electrochemical oxidizing reactions at the surface of metals.
-
(5)
The in-core redox water potential was observed to be approximately +0.1 V higher than the out-of core region. The high redox water potential attracts conduction electrons through piping into the redox water, reducing dissolved oxygen (DO) and H2O2 through two reaction channels.
-
(6)
The potential in the out-of-core region should be determined by mixing these two contributions.
-
(7)
Although the out-of-core region is lumped into a single volume of the redox water to be consistent with the point model, the actual potential changes gradually due to the electrochemical kinetic effects of oxidizing H2O2 to O2when flowing through the piping.
This mechanism was discovered while attempting to reconstruct the potential differences observed in the two in-pile test loops; NRI-Rez in the Czech Republic (Kysela et al., 2001, Zmikto, 2003, Kaselam et al., 1998) and INCA Loop in Sweden (Takiguchi et al., 2004, Takiguchi et al., 2000) both provided “spectroscopic” data by largely changing the concentrations of dissolved oxygen/hydrogen, respectively. Through modeling studies which will be introduced in Chapters 3–4, it was determined that there should be an electrochemical kinetic mechanism oxidizing the stable molecular products in the out-of-core region with the circulating reactor water in metallic loops. Mutually counter-cell reactions are occurring at these two regions with the “differential radiation cell (DRC)” configuration.
2.2. Differential radiation cell (DRC) and radiation-induced electrolytic (RIE) corrosion
2.2.1. Experimental evidences on DRC
For the BWR water chemistry sphere there are several experimental evidence demonstrating that irradiation can induce electrochemical potential shifts, which is an interesting mechanism specific to the nuclear reactor environment. Although existence of this effect is well established through several independent experiments assessed by the author (Saji, 2009), there are very few “controlled” experiments which are robust enough to withstand further theoretical studies. Most of these experiments do not model the actual reactor configuration, such as dose rate, temperature, flow velocity and circulation, nor the concentration of solute species. In addition, these experiments do not simulate the RIE configuration with the cathodic irradiated- and the out-of-flux anodic regions which are connected through the metallic piping.
More recently the redox potential differences were measured between two regions, one with an electrode installed at an in-flux region and another at an out-of-flux region during which dissolved oxygen levels were altered as an experimental parameter to cover a wide range of concentrations. These results were obtained by the Nuclear Research Institute (NRI) in Rez, Czech Republic (Kysela et al., 2001, Kaselam et al., 1998, Zmikto, 2003). Their report demonstrated the existence of an approximate +100 mV ECP difference between the in-flux ECP electrode and those at the out-of-flux regions. In Chapter 4 of this report, the author demonstrates that this potential difference can be reproduced reasonably well from the radiation chemical calculation. The ECP electrode system contains a platinum electrode and wiring that enables one to measure the potential on the experimental environment, which is then compared with the reference Ag/AgCl electrodes. The platinum electrodes were placed on the main piping at a temperature of 288 °C with a flow velocity of ∼2 m/s and an oxygen concentration that varied between 0 and 700 ppb. Although details have yet to be reported, this would be direct evidence that radiation can induce a potential difference in the cooling water with a wide range of dissolved oxygen concentrations. Even in this test the absorbed dose rate at the measured point is substantially smaller than the actual BWR core, therefore theoretical reconstruction of the experimental results are indispensable to extrapolate the currently available experimental results to the actual BWR cores.
Also in recent studies with the goal of introducing an optimized DH level in the PWRs, the Studvik Nuclear team in conjunction with the Japan Atomic Power Company’s (JAPC) team performed in-pile experiments as well as model calculations for the radiolysis of the PWR-simulated primary circuit at the JAPC’s Unit 2 of the Tsuruga PWR (Takiguchi et al., 2000, Takiguchi et al., 2004). They confirmed that the in-pile measurements of stainless steel ECP indicated a potential which was high and sensitive to the DH level. The ECP of stainless steel against the platinum (Pt) electrode indicated −300 mV SHE although the contemplated potential was −700 mV flat. Their experiment for the ECP of stainless steel, as measured against the Pt electrode, was converted to the SHE scale which took into account pH, temperature and DH (Molander, 2008). However, the out-of-core ECP was not measured together with the in-core potential (i.e., no electrode was available at the inlet of the in-vessel rig). In addition, although degassed and purified water is used, the water is not circulated in their experiment (Christensen et al., 1996).
Prior to their experiments the ECP was believed to be flat and close to the potential induced by the DH at approximately −700 mV SHE. This potential was confirmed through measurement in an actual PWR (Molander et al., 1986). However the experimentally observed jump in potential at the lower end of DH implies that the degassing might have been insufficient in the INCA Loop with its once through feed water system design. Since a large increase in the ECP was once reported below the DH concentration of 2 N-cc/kg, additional data on the out-of-core ECP are awaited to confirm whether the reported peculiar phenomenon is due to the insufficient degassing effect.
2.2.2. Mechanism of DRC inducing RIE
With DRC severe radiation-induced electrolytic corrosion (RIE) is anticipated. These two phenomena are like two sides of a coin and for this reason the author has attempted to theoretically reconstruct the experimentally observed redox potential differences by developing the basic theory for both PWR and BWR water chemistry environments which will be introduced in Chapter 2. This phenomenon can be qualitatively explained for the PWR case as follows:
The basic radiation chemistry process is well established such as in the textbook by (Spinks and Woods, 1990). During the chemical stage (10−12–10−6 s) of water radiolysis, the species react in the tracks and then diffuse in the solution. They can thus react with each other as well as with the surrounding molecules (in the solute). The track of the particles expands due to the diffusion of radicals and their subsequent chemical reactions. Recombination becomes unimportant after an order of 1 μs for low-LET radiation. A simplified interaction of radiation with water can be expressed in:(1)
The last two ions are seldom measured as radiation chemical species, being masked by abundant ions produced through the electrolytic dissociation of water (i.e., Ostwald’s law which determines pH).
Among other species only the hydrated electron, e−aq, is a charged species since the concentration of HO2/O2− can be insignificant in water with moderate pH. By performing a combination of radiation chemistry with electrochemistry calculations, the hydrated electrons, e−aq, reacting with stable molecules are found to be primarily responsible for inducing a large portion of the potential differences both in the PWR and BWR water chemistry environments (Saji, 2009, Saji, 2010).
Although the exact mechanism is not known an electrochemical potential difference of 0.1 V (BWR-NWC), ∼0.3 V (PWR) is observed in the core region which will be introduced in greater detail in Chapter 4. This potential difference is measured through in-pile tests with an equivalent hydrogen standard electrode which is formulated by dosing the primary water with hydrogen gasin the INCA Loop and a standard electrode that is inserted in the flux region. In the measurement, the potential of the electrode should be raised by 0.1/0.3 V, for BWR-NWC/PWR respectively, by applying an external battery to cope with the redox potential. Note that the hydrated electrons, with their short lifetime, do not flow directly to the electrode. Rather the hydrated electrons generated in the core region participate in raising the redox potential of the water and induce an electron current from the metallic wall to the reactor water in the hydrogen water chemistry through electrolytic reaction at the surface of metal:(2)
A supply of hydrogen ions are limitless due to the overwhelming large concentration of water molecules which dissociate into H+ and OH−.
With the in-core concentrations larger than those of the out-of-core region, DH is reverted back to the hydrogen ion at the out-of-core region through the oxidation of hydrogen at the surface of metals:(3)
Here the excess e−cathode is transported back to the cathodic reactor core region through piping thereby closing the electron conducting circuit. Eq. (3) is the counter cell reaction of Eq. (2). These two mutually counter-cell reactions are very important in maintaining the overall material balance of the loop with circulating water by means of radiation difference. The existence of the DRC is illustrated in Fig. 3 which demonstrates that the hydrogen concentration at the outlet of the in-core region is higher than that at the inlet of the in-core region. The graph demonstrates the electrode kinetic mechanism which should decompose the DH that would be expected at the out of the core region through oxidation where the necessary electrons are removed at the electrode (i.e., metallic wall).
 Fig. 3. Comparison of DH of in- and out-flow from the in-core region. (Note) This figure demonstrates that due to radiation, the out-flowing DH (solid line) is higher than the in-flow concentration (broken line). The former must be decomposed to the in-flow concentration in the out-of-core region. The overall mechanism is illustrated in the INCA Loop case as presented in Fig. 2.
Fig. 3. Comparison of DH of in- and out-flow from the in-core region. (Note) This figure demonstrates that due to radiation, the out-flowing DH (solid line) is higher than the in-flow concentration (broken line). The former must be decomposed to the in-flow concentration in the out-of-core region. The overall mechanism is illustrated in the INCA Loop case as presented in Fig. 2.The set of electrochemical reactions between the in- and out-of-core regions result in a net effect of the conduction electrons (i.e., long-cell current) flowing from the cathodic surface (e.g., fuel cladding) into the redox water. Fig. 4illustrates the “specific current” (electron current flowing into the redox water per unit volume) calculated by using the same model used in Fig. 3. The current is estimated from the net rate of DH flowing into the core region minus the out flow from the out-of-core region with the ‘normalized’ volumetric flow rate , which represents the flow rate of the coolant in the reactor core in a point model, namely:(4)where n is the number of electrons transferred per molecule, F is the Faraday constant, Cox is the concentration expressed in mol/dm3 for species ox in-core region , which applies to the stable molecular species such as H2, O2 and H2O2. In Eq. (4), the sign of io is assigned positive/negative for PWR/BWR-NWC respectively.
 Fig. 4. Estimated equivalent electron current density flowing through piping. (Note) This figure shows the electron current transported from the out-of-core anodic region to the in-core cathodic region flowing in metallic piping an ampere per liter for the PWR reactor water environment.
Fig. 4. Estimated equivalent electron current density flowing through piping. (Note) This figure shows the electron current transported from the out-of-core anodic region to the in-core cathodic region flowing in metallic piping an ampere per liter for the PWR reactor water environment.The specific current is estimated by assuming that there is no current density limit due to the over potentials induced such as in the hydrogen rich layer which likely exists at the cathode surface. Actually the in-pile test results indicate that there should be a significant depletion in DH below approximately 2 N-cc/L as discussed in Section 5.1. By limiting the specific current density at less than 8.4 A/L below DH = 10 N-cc/L, the author confirmed that the deviation is not due to the current density limiting effect. Although the long-cell current has never been measured in the reactor configuration, however, analogous “long-cell current” has been measured in soil corrosion by E. Schaschl and G. A. Marsh. Similar experimental verification with DRC is highly awaited.
A similar mechanism also exists in the NRI-Rez in BWR-NWC case as shown in Fig. 5. In this case the oxygen concentration in the outlet of the reactor core region is substantially lower than that of the inlet oxygen concentration, indicating that the irradiation effects should enhance the reduction reactions in-core. The concentration difference should be supplemented by the counter cell reactions of oxidizing hydrogen peroxide (Eq. (5)) at the out-of-core region as shown in Fig. 5.(5)
 Fig. 5. Comparison of DO and H2O2 concentration between in- and out-flow from the in-core region. (Note) This figure illustrates that the out-flowing DO (solid line) is lower than the in-flow concentration (broken line) due to radiation. The DO concentration must be increased to the in-flow concentration in the out-of-core region through the counter cell reactions involving both O2and H2O2. The concentration of H2O2 is plotted with two horizontally broken and dotted lines. The reaction channels are illustrated in the NRI-Rez Loop case of Fig. 2.
Fig. 5. Comparison of DO and H2O2 concentration between in- and out-flow from the in-core region. (Note) This figure illustrates that the out-flowing DO (solid line) is lower than the in-flow concentration (broken line) due to radiation. The DO concentration must be increased to the in-flow concentration in the out-of-core region through the counter cell reactions involving both O2and H2O2. The concentration of H2O2 is plotted with two horizontally broken and dotted lines. The reaction channels are illustrated in the NRI-Rez Loop case of Fig. 2.Fig. 6 illustrates the “specific current” (electric current flowing into the redox water per unit volume) calculated from Fig. 5 assuming there is no current density limit due to over potentials. Actually the in-pile test results indicate that there appears be a significant increase in H2O2 in the region of DO below approximately 1 ppb as discussed in Section 5.2.
 Fig. 6. “Specific current” in the NRI-Rez Loop’s BWR-NWC. (Note) This figure illustrates that “the specific current” io is flowing into the redox water from the metallic surface in A/L. In the test a significant increase in H2O2 is identified below DO < 1 ppb due to a limiting current density in the in-pile test results, although the results of the numerical computation are suppressed due to the truncation errors with Eq. (4) at such a low concentration.
Fig. 6. “Specific current” in the NRI-Rez Loop’s BWR-NWC. (Note) This figure illustrates that “the specific current” io is flowing into the redox water from the metallic surface in A/L. In the test a significant increase in H2O2 is identified below DO < 1 ppb due to a limiting current density in the in-pile test results, although the results of the numerical computation are suppressed due to the truncation errors with Eq. (4) at such a low concentration.The reactor water with the lower in-core DO concentration, lower than the inlet DO concentration, is flowing out of the core region where the redox water is oxidized at the out-of-core region. This oxidization process should take place gradually with distance from the in-core region. Fig. 7 demonstrates that this electrochemical kinetic mechanism taking place at the outlet piping of the NRI-Rez BWR-2 loop (Kaselam et al., 1998). In Fig. 7 the variation of DO concentrations are plotted against different inlet DO concentration with distance from the core outlet used as a parameter. Sixty meters from the outlet of the in-core region represents “K3 out-of-core channel” which is used as the representative out-of-core measurement point in this report. Although the total length of the loop is 95 m, the DO concentration should be close to the inlet concentration of the core region. This distance variation is due to the electrochemical decomposition of H2O2 whose concentration changes with distance which is also demonstrated in Fig 8.
 Fig. 7. Variation of DO concentration with respect to distance from the core. (Note) The variation of DO concentration at the outlet is assumed to be equal to that of the in-core concentration while changing the inlet DO concentration, with the distance from the core as a parameter. The DO concentration along the piping changes due to the electrochemical decomposition of H2O2. When the dissolved H2O2 is depleted the water is decomposed as per 2H2O → O2(aq) + 4 H+ + 4e−cathode.
Fig. 7. Variation of DO concentration with respect to distance from the core. (Note) The variation of DO concentration at the outlet is assumed to be equal to that of the in-core concentration while changing the inlet DO concentration, with the distance from the core as a parameter. The DO concentration along the piping changes due to the electrochemical decomposition of H2O2. When the dissolved H2O2 is depleted the water is decomposed as per 2H2O → O2(aq) + 4 H+ + 4e−cathode. Fig. 8. Variation of H2O2 concentration with distance. (Note) H2O2 Concentration is little affected by water chemistry control through the DO control, however, it depends strongly on the distance from the core region through the electrochemical decomposition. Before reaching the inlet of the in-core region, its concentration is almost zero. The H2O2 concentration is fitted with the experimental results of the out of core potential which indicates Eo(total) = Eo(O2) + 0.289Eo(H2O2) where the weight of the H2O2 concentration (i.e. 0.289) is obtained from the theoretical potential of Eo(O2) and Eo(H2O2) as explained in Section 6.2.
Fig. 8. Variation of H2O2 concentration with distance. (Note) H2O2 Concentration is little affected by water chemistry control through the DO control, however, it depends strongly on the distance from the core region through the electrochemical decomposition. Before reaching the inlet of the in-core region, its concentration is almost zero. The H2O2 concentration is fitted with the experimental results of the out of core potential which indicates Eo(total) = Eo(O2) + 0.289Eo(H2O2) where the weight of the H2O2 concentration (i.e. 0.289) is obtained from the theoretical potential of Eo(O2) and Eo(H2O2) as explained in Section 6.2.The corresponding H2O2 concentration behind Fig. 7 is calculated as shown in Fig. 8. Since H2O2 is solely produced with irradiation, it is independent from the variation in DO concentration at the inlet but changes with distance from the core region.
2.2.3. Prevention of RIE corrosion
The “electrolytic corrosion” is often confused with galvanic corrosion. While the latter is driven by the difference in the corrosion potential between two metals, the electrolytic corrosion is driven by the external sources of EMF (electromotive force). The RIE corrosion is a special case of electrolytic corrosion in which the EMF is induced between the irradiated- and un-irradiated portions of the water in metallic loops. Thus the selection of materials is of a little effect in reducing EMF.
In spite of the complexities pertaining to the radiation- and electrochemical processes involved in describing the in-core chemistry as developed in Chapters 2–4, the prevention of this mode of corrosion requires only provisions that break the continuity of the electrical circuit. As a matter of fact, prior to the US developing the theory of LGCA corrosion during the 1960s, in 1952, H. W. Wahlquist demonstrated the remarkable effectiveness of electrical insulation in the mitigation of corrosion issues in a fossil power plant (Wahlquist, 1952). Walquist, an electrical engineer, visualized the corrosion problems encountered in steam-electric power plants as, in effect, a huge short-circuited battery with current discharging to the cooling water from the interior surfaces of the water-box, and then collecting on the tube-sheet and tube ends. Three surface condensers constructed for a new steam-electric station were provided with electrical insulation between the cast-iron water-box flange bolts, and also the flanges for the auxiliary piping. The insulation successfully stopped corrosion. Similar provisions should be feasible for water-cooled reactors such as by applying a plasma spray coating to fuel assemblies at locations in direct metallic contact with the in-core structures.
2.3. Electrolytic phenomena and aggravation of anodic corrosion activities
The RIE induces a redox potential difference between the reactor core and the out-of-core regions. With the potential difference it is a natural approach to anticipate the electrolysis phenomena such as the decomposition of reactor water into hydrogen and oxygen. Such an effect is adequately devised during normal operation through the removal of radiolysis products and appropriate water chemistry control. However, the electrolytic decomposition of water would have been uncontrollable during the recent severe accident. In particular, the mysterious hydrogen explosion at the Spent Fuel Pool in Unit 4 of Fukushima Daiichi can be explained through this mechanism (Saji, 2015a, Saji, 2016) as summarized in Annex B.
Since the RIE mechanism was found by extending the LGCA corrosion hypothesis (Saji, 2015b), the “effects of long cell on local cells” induces suppression of local cell corrosion activities at the cathodic regions and in general aggravates local cell activities in the anodic regions as demonstrated in soil corrosion. One of the most visible cathodic activities is the deposition of CRUD on fuel rods assessed in the author’s previous report (Saji, 2015b) as summarized in Annex C. Here “CRUD” is an acronym of Chalk River Unidentified Deposits, first identified while developing the Mark I prototype reactor for the first nuclear powered submarine, the Nautilus. To verify that the reactor’s fuel elements would not corrode, samples were placed in a research reactor located at Chalk River, Canada. After several months of irradiation, the fuel elements were covered in yellowish deposits. Although the deposition of corrosion products on fuel rods are universal problems in LWRs, its root cause has not been scientifically clarified.
The anodic reactions induce various corrosion phenomena, aggravated by the RIE mechanism. This should result in the unexpected aging of components even when manufactured with “corrosion free” materials combined with stringent water chemistry control. One of the typical examples is unexpected early failure of the BWR’s shroud as summarized in Annex D. Knowing these electrolytic phenomena and the aggravation of anodic corrosion activities, the following discussion focuses on elucidating the mechanism of “radiation-induced electrolytic” corrosion in LWRs.
2.3.1. In-core chemistry
Since the mechanism is closely related with “in-core” chemistry which has been considered as a black box that cannot take an accurate measurement of in-core chemistry parameters such as pH, potential and concentration of radical- and stable molecular species. For example until recent years, PWRs’ ECP has been assumed unchanged from that of ECP determined by dissolved hydrogen concentrations. However, the author believes it is now becoming possible to theoretically estimate in-core chemistry parameters thanks to the recent development of radiation chemistry which measured reaction sets, rate constants and g-values with pulsed radiolysis performed with high radiation pulses. In view of this notion, the chemical environment of a water cooled reactor is briefly reviewed before discussing radiation chemical formalism used in estimating the in-core chemistry.
In LWRs the reactor water is circulated through the reactor core region and nuclear heating generated there is removed either by the steam generators in PWRs or through boiling heat transfer at the reactor vessel in BWRs. In the PWRs’ water chemistry configuration, the reactor water is dosed with hydrogen and boric acid with an alkaline neutralizer such as LiOH to adjust pH. The dosing of the hydrogen is required for PWRs to prevent the net breakdown of the water (i.e., water radiolysis) and to suppress its concentration of oxidative radicals such as OH. Whereas it is not possible to prevent the generation of hydrogen in BWRs due to the “unclosed configuration” (Spinks and Woods, 1990) with a steam separator space at the top, the radiological generated hydrogen is extracted from the turbine condenser vacuum as residual gas. Since the oxidative radicals are not suppressed in BWRs, their reactor water can be characterized as “oxygen water chemistry” although in recent years a small amount of hydrogen is dosed to mitigate the corrosion phenomena.
Using the published in-pile test results as well as modeling studies as summarized in this review, it is now established that the electrochemical potential of the reactor water at the core region is more positive than the out-of-core regions in both PWRs and BWRs. These in-pile test results were published rather recently as summarized in Section 2.2.1, although comprehensive experimental parameters have not been published since these data were obtained as a by-product of in-pile material tests and water chemistry optimization. Further review of these results will be discussed in Chapter 4. The higher redox potential in the core region is induced by the strong radiation of gammas and recoil protons as well as alpha particles in PWRs. These effects are incorporated in the author’s analysis through g-values, in which further discussion should be referred to the reference (Elliot and Bartels, 2009). These potential differences induce an electric current flowing through piping resulting in RIE effects as explained in Section 2.2.3.
The redox potential of water in the out-of-core region is calculated by assuming an equilibrium state (i.e., using Nernst equation). However the deviation from the equilibrium state occurs at the outlet of core region with a gradually reducing potential with distance due to an electrochemical kinetic effect. These two regions coupled as a pair of cathodic- and anodic half-cells by conducting the electric current through piping (named “long-cell-current”). With circulation of the reactor water the radiologically generated molecular species are transported to the out-of-core anodic region. Although they are charge neutral they carry valence electrons. These charge neutral species are oxidized at the anodic out-of-core region by releasing electrons to the metallic wall in effect closing both the material balance and electron balance circuits as illustrated in Fig. 2. Note that both a material and electron balance should be established.
If the RIE reaction is induced, significant differences in ECPs must be observed between the anodic sites where the dissolution of metal (i.e. corrosion) is taking place, and cathodic sites where deposition (also called sedimentation) of corrosion products is often observed. Although there is no appreciable difference in voltage anticipated along the metallic structure, the redox potential difference is induced between the local electrolyte in contact with the anodic metal surface (an anodic half-cell) and the cathodic counterpart. Between these two sites a significant electric current (i.e. “long-cell current”) should be identified although it has not been measured to date in the reactor configuration2.
2.3.2. Radiation-induced environmental SCC
Laboratory and field data relating to ‘environmental corrosion’ demonstrate that the accelerated inter-granular stress corrosion cracking of stainless steels and nickel-base alloys can be the result of long term exposure3 to the high energy neutron and gamma radiation that exists in the core structures of LWRs (ANL, 2010). Radiation can exacerbate specific aspects of cracking (e.g. IGSCC) susceptibility via its effects on ‘material micro-chemistry’ (e.g., radiation induced segregation), water chemistry (e.g., radiolysis) and stress (e.g., radiation induced creep and hardening) (Andresen et al., 1989). More recently, Mi Wang has published an overview of irradiated assisted corrosion of stainless steel in LWRs by focusing on radiolysis and corrosion (Wang, 2013). He stresses the role of the protective passive film consisting of a chromium-rich continuous inner layer, covered with an iron rich porous outer layer. The existence of this protective layer influences PWR water chemistry, temperature, stress, pH and therefore can cause a variation in the properties of the oxide film. However, combining all these influences together, as in real PWR conditions, may result in stress corrosion cracking. In the context of his conclusion, SCC is un-preventable with so many complex factors influencing the durability of the protective film in the PWR environment.
2.3.3. De-passivation with RIE
However, the RIE mechanism explains the loss of the protective film (i.e., de-passivation) since an electric current is induced at the anodic surface of metals. As early as 1949, Italian scientist G. Petracchi demonstrated that electrochemical effects significantly influence erosion rates (Petraccchi, 1949, Saji, 2006). He investigated whether a primary influence of corrosion from ‘cavitation’ could have attributed to the anodic reaction currents, either from external causes (e.g., stray currents or galvanic couplings) or from internal causes, including the development of micro-pits originating from localized mechanical stress. He performed two series of experiments, i.e., fatigue and corrosion tests, as well as flow nozzle ‘cavitation’ (FAC) tests. For the latter, he constructed a flow nozzle with two specimens kept under external electrical potential.
Upon inducing as low as 0.1 mA/cm2 of the positive current, the erosion rates reported were drastically different. The extensive weight loss observed by applying the positive potential to the specimen indicates that the protective oxide film is lost. By reversing the polarity, Petracchi observed that the surface was covered with a film exhibiting light-interference reflection bands, which is indicative of the presence of a protective film. One of Petracchi’s experimental results is reproduced in Fig. 9. A slight weight increase meant no erosion was observed by reversing the polarity of the potential. These electrochemical factors have been overlooked in FAC studies to date. Based on his experiments carried out on various metal, he demonstrated that very weak currents exerted cathodic protection or anodic corrosion and were in fact dominant in corrosion from ‘cavitation.’ Unfortunately, his experiment did not seize the attention of corrosion scientists and no systematic experiments have been performed to the author’s knowledge.
 Fig. 9. Results of the ‘cavitaion’ (FAC) test with electrical potential. De-passivation of iron specimen occurs when positive potentials are applied. When the potential is reversed, FAC completely ceases. Reproduced from reference (Petraccchi, 1949).
Fig. 9. Results of the ‘cavitaion’ (FAC) test with electrical potential. De-passivation of iron specimen occurs when positive potentials are applied. When the potential is reversed, FAC completely ceases. Reproduced from reference (Petraccchi, 1949).2.3.4. IASCC as an example of RIE corrosion
Exacerbation of IGSCC in the radiation field is called IASCC. In general, it can be reproduced on the materials irradiated over a certain threshold fluence level of fast neutrons followed by the post-irradiation fatigue examinations (PIEs) (ANL, 2010). However the reproduced IASCC by PIEs must be carefully compared with the actual IASCC in LWRs, because the latter occurs near the core region under the simultaneous effects of radiation, stress and a high temperature water environment. To confirm the effect of synergy, Japan Atomic Energy Agency (JAEA) developed the test technique to carry out in-pile IASCC tests at the Japan Materials Testing Reactor (JMTR) (Tsukada, 2006). The test results indicate that the synergy effects of neutron/γ-gamma radiation and stress/water environment on SCC growth rate was considered to be minor within the current test conditions (Miwa et al., 2007). Further studies are currently awaiting the restart of JMTR which was shutdown in 2006 for refurbishment. Further discussions on this issue are summarized in Annex D.
3. Development of the basic theory of the RIE phenomena
Several steps are required for the assessment of the RIE phenomena as illustrated in Fig. 10. Note that the overall approach can be divided into two groups, material balance assessment and potentiometric assessment. Although the material balance results are very useful as demonstrated in Fig. 3, Fig. 4 as well as in Annex B, only limited experimental data related to molecular species are available. Since the experimental data are on potential differences induced by radiation it is necessary to proceed to the bottom half potentiometry steps in Fig. 10 for further verification. Since it is necessary to introduce several physical chemistry parameters, some additional uncertainties were unavoidable.
 Fig. 10. Flow chart of development and verification of the basic theory of the RIE phenomena. Verification starts with data pertaining to the plant (e.g., plants’ radiation chemical and electrochemical configuration data) and that related to a radiation chemical reaction set, rate constants and G-value. Going through two steps of material balance assessment (radiation chemical material balance equation integrated with Faraday’s law and QSSA), the intermediate results are concentrations of radical- and stable molecular species. Since the experimental data are related to the potential differences it is necessary to proceed to the potentiometry steps at the bottom half of Fig. 10 for further verification.
Fig. 10. Flow chart of development and verification of the basic theory of the RIE phenomena. Verification starts with data pertaining to the plant (e.g., plants’ radiation chemical and electrochemical configuration data) and that related to a radiation chemical reaction set, rate constants and G-value. Going through two steps of material balance assessment (radiation chemical material balance equation integrated with Faraday’s law and QSSA), the intermediate results are concentrations of radical- and stable molecular species. Since the experimental data are related to the potential differences it is necessary to proceed to the potentiometry steps at the bottom half of Fig. 10 for further verification.3.1. Role of hydrated electrons (e−aq)
As explained in the Introduction, the RIE effects should be aggravating the anodic corrosion activities in LWRs. This mechanism was identified with the potential differences observed in two in-pile test loops; the NRI-Rez in the Czech Republic and INCA Loop in Sweden. These results indicate that the in-core redox potentials are approximately 0.1/0.3 volt higher, in BWR(NWC)/PWR water chemistry respectively, compared to that of the out-of-core regions.
The author has demonstrated that such a potential difference is induced through radiation chemical reactions including annihilation of hydrated electrons generated under radiation (Saji, 2010, Saji, 2013). However the hydrated electrons e−aq, with their short lifetime, do not flow directly to the electrode. Rather the hydrated electrons generated in the core region participate in raising the redox potential of the water and induce an electron current from the metallic wall to the water. The higher redox potential attracts both corrosion electrons as well as electrons generated from the anodic reactionof the reactor water through Eq. (3) to the core region with the counter cell reaction, Eq. (2), therefore the aggravation of the corrosion phenomena should be induced.
3.2. Radiation chemical material balance assessment
3.2.1. Integration of radiation- and electro-chemistry processes
The “chemical” form of the reaction for radiation with water should be further deployed in characterizing the radiation chemistry process in reactor water by adapting NPP specific conditions, namely:
-
(1)
The reactor water, in a high purity state, is circulated within the metallic coolant loops.
-
(2)
Solute species are added or removed through water chemistry control, such as by dosing with hydrogen/ammonia4 in PWR/VVER respectively and oxygen in BWR (NWC)5 or RBMK environments. Therefore the following reactions occur in the reactor water environment along the loop.
Along the loop, the following processes are involved.(6)
The basic difficulty in Eq. (6) is in the fact that the generation or removal of species through electrolysis (i.e. Faraday’s Law) is not included, thus the overall electrochemical material balance is not established in the loop. The stable molecular species should be in a state of equilibrium in which the imbalance in generation and removal rates should be compensated through the electrical current induced by the RIE.
3.2.2. Electrolytic generation of H2 and O2 at electrodes
The observed deviation of the dissolved hydrogen and oxygen concentrations were shown to be induced by a generation of hydrogen (in PWRs and VVERs) and oxygen (in BWR-NWC and RBMK) through the RIE processes. The author has demonstrated that the deviations in concentrations of these molecules are equivalent to the concentrations induced through an exchange current density,6io, through the electrochemical transfer processes.
The most common electrochemical transfer reactions occurring on the surface of the electrodes for PWR/VVER and BWR(NWC)/RBMK include;(7)(8)(9)(10)
Note that the last two electrochemical reactions designate the “doubly unstable domain” in which the hydrogen peroxide decomposes into water and oxygen, only in this potential domain can hydrogen peroxide decompose chemically into water and oxygen (Pourbaix, 1966). Since the standard potential of H2O2through Eq. (10) is very large (approx. 1.7V), this reaction channel can be ignored in the case of LWRs. These reactions are generally expressed as:(11)where Ox is the oxidized- and Rx is the reduced species and n is the electric charge transferred through the particular reaction involved.
3.2.3. Radiation-chemical processes without electrochemical effects
In NPPs when the electrochemical processes are NOT included, the basic time dependent material balance equation for radiation chemical species can be described as:(12)where Ci is the concentration expressed in mol/dm3 for species Xi, g (Xi), the g-value in mol/J for species Xi, is the dose rate in Gy/s, kis is the rate constant between species Xi and Xs in dm3 mol−1 s−1. The rate constants kiqr (represented for the second order reactions, in dm3 mol−1 s−1) between species Xq and Xr, have non-zero values only when the reactant species is Xi in the quasi second order reaction. The ‘normalized’ volumetric flow rate represents the flow rate of the coolant in the reactor core within a point model and is the concentration of the species-i, in the incoming flow, which applies to the stable molecular species such as H2, O2 and H2O2.
In the steady state the concentration of long-lived species can be calculated with the following expression as shown in Eq. (13) for long-lived species and (13′) for the short-lived radical species:(13)(13′)
Here the in-flow of radical species are ignored due to its extremely short lifetime.
The simplification with these two sets of equations (13), (13′) is called “improved” QSSA.7 It is widely used in the integration of stiff ordinary differential equations8 arising from the photolysis effects in the atmospheric chemistry arena (Jay, 1995). When the equation dealing with long-lived molecular species is not separated from the equation for short-lived radical species, such a simplification is called the “plain” (i.e., original) QSSA, and may result in erroneous solutions as discussed in reference (Farrow and Edelson, 1974). Due to the difficulties in solving the stiff nature of Eq. (12), the established computer codes with Gear’s numerical integration algorism have been more commonly used among radiation chemistry specialists (Gear et al., 2002).
3.2.4. Radiation-chemical processes with electrochemical effects
The basic time dependent material balance equation for Ox in the radiation chemical environment can be described in Eq. (14), when the electrochemical processes (i.e., Faraday’s law) are included in a “point model” (where the flux region is lumped together into a single water volume Vo), as:(14)where the newly introduced electrolysis parameters are the cathode current icathode per unit volume of the redox water and the Faraday constant F. In the last term of Eq. (14), representing the electrolytic generation/removal of Ox, the sign of the icathode is assigned as positive for the forward9 reaction (reaction to the right) in Eq. (11).
It is important to point out that the cathode current can be introduced by external systems typically through RIE, coupled with the water chemistry configuration outside of the radiation chemical system under consideration. Further discussion will be made in Chapter 5 Discussions. In the steady state, the concentration can be calculated in the following “improved” QSSA expression for molecular species:(15)(13′)
In the “improved” QSSA, the concentration of the oxidized species should be calculated from Eq. (15) by including the electrolytic reaction, although Eq. (13′)can calculate concentrations of radical species.
When the reactor water is in a state of equilibrium with circulation, Eq. (15)leads to a simple relationship between icathode and the concentration of radiation chemical species:
(16)
This equation demonstrates that the concentration of Ox removed through electrolysis amounts to the radiation-induced excess Ox (the top two terms in the bracket), which is generated through radiation but insufficiently removed by it (the lower term). Note that this evidence is made without using the particular radiation chemistry parameters although the numerical value of the cathode current (i.e., “long cell” current) can be estimated by applying these radiation chemistry parameters.
Eq. (17), the “Butler-Volmer equation” (Shoesmith, 1987), is an electrochemical representation of the cathode current(17)where io is called exchange current (i.e., the specific current at an equilibrium state),10 R is the gas constant, T is the absolute temperature and β is the symmetry coefficient taken to be close to β = 0.5. The term η is the over-potential at the core and is a measure of how far the cathode reaction is from equilibrium.When the surface concentration is close to that of bulk water and |η| is large, the Butlar-Volmer equation can be simplified to the following Tarfel equation:(18)
Therefore with the Tarfel approximation the cathodic over-potential in the flux region can be estimated by:(19)
which should be the potential difference induced by radiation. The over-potential is one of the important electrochemical kinetic parameters obtained through experiment; however it can be estimated from the radiation chemistry calculation as introduced in this section when experimental io is known.
For a corrosion study which deals with anodic reactions; M → Mn+ + ne−;another anodic Butler-Volmer equation should be introduced where the over-potential should be determined by Eq. (19) from the water chemistry specifications. When the cathode current is not sufficiently supplied from the anode through the reaction Eq. (3), the deficiency can be supplied through accelerated corrosion.
3.3. Potentiometric assessment
With the estimation of the equilibrium concentrations of both oxidized molecular and ionic species as well as radical species by way of Eqs. (15), (13′), it is now ready to proceed into the potentiometric assessment. This procedure is necessary for example, to estimate the in-core ECP as well as further verification by applying the currently available experimental results of the in-pile tests. However, it was found that converting the concentrations of molecular and radical species to electrochemical potentials is not well established. As a matter of fact a more recent review report by the winners of the 2011 IUPAC Prize stated; “despite extensive efforts devoted to obtaining these values, absolute ion solvation enthalpies and energies have not been measured directly” (William and William, 2011). Even though a vast amount of physical chemistry reports on water have been published, this situation changes greatly when the effects of radiation are involved due to the anticipated difficulties in performing such experiments under such conditions. However the author believes that an integration of the radiation chemical reaction sets, rate constants and g-values should elucidate many of the unknowns.
One of the most practical approaches for converting the concentrations of molecular and radical species to electrochemical potentials should be a Nernst-like equation by using the “formal potential” which replaces the standard potential to include some small corrections to the logarithm term. (IUPAC, 1997). is the potential that is actually measured in an electrochemical cell in a non-standard condition where the activities of Ox and Red are affected by radiation.
3.3.1. Applicability and modification of the Nernst equation for “nuclear” corrosion studies
Although the author’s previous publications have demonstrated the viability of this new approach, his efforts were only partially successful in providing the initial objective since it was necessary to introduce a fitting parameter to reproduce the experimental potential values in effect using the formal (conditional) potential11 as the fitting parameter. This observation is closely related with thermodynamic difficulties of a standard potential for hydrated electrons as discussed by Conway and MacKinnon (1970). According to their discussion, the standard potential for the hydrated electrons e−aq on the hydrogen scale has been calculated from kinetic data to be ca. −2.67 V yet hydrogen evolves easily on the more catalytic metals at appreciable rates already at 0.1–0.2 V cathodic to the reversible H2 potential. These difficulties can be removed in the framework of RIE, since Conway and MacKinnon are discussing “open-circuit corrosion processes” whereas RIE is a closed-circuit corrosion process. The currently used standard electrode potential value of 2.86 V (Spinks and Woods, 1990) is too large to explain the observed potential differences.
Some scientists argued against the author’s reliance on the Nernst equation:(20)where Eo is the equilibrium electrode potential of the irradiated water, is the standard equilibrium potential of the water typically containing DH or DO, R is the gas constant, T is the absolute temperature, n is the number of electrons transferred in the half-cell reaction, F is the Faraday constant and (OX) and (RED) are the activities (or fugacity) of the oxidized and reduced species, respectively. In general the Nernst equation applies to an electrode process that is thermodynamically or electrochemically equilibrium since “there is always an equilibrium condition at the base of the discussion of any kinetic process” as pointed out by Bockris et al. (2000).
Assuming a state of equilibrium, the author tried to develop a formalism by extending the Nernst equation to deal with the observed redox potential differences since the objective is to apply it to corrosion studies. In corrosion science, it is well established that Pourbaix’s Atlas of Electrochemical Equilibria (Pourbaix, 1966) provides information about the stability of metals as functions of pH and potential E. It provides information on the three states of metals:
-
•
Corrosion: active state
-
•
Passivity: forming passive layers inhibiting the corrosion process on the surface of the metal
-
•
Immunity: thermodynamic stability
However, in the RIE configuration the passive films can be removed (i.e., de-passivation) when an external positive potential is applied as demonstrated by Petracchi. By losing the protective oxide film, the corrosion rate was accelerated by several orders of magnitude. When the potential was reversed, corrosion was completely ceased and a passive film was confirmed (Petraccchi, 1949). Therefore passivity may not guarantee the protection of metals in the RIE configuration. This de-passivation mechanism is very important for structural materials used in LWRs since they are almost always exposed to the reactor water in the passive region.
Pourbaix’s diagrams are constructed by applying the Nernst equation as it is derived entirely from chemical thermodynamics, his atlas can also be used to determine which species (metals or alloys) are thermodynamically stable. As such, Pourbaix’s atlas is indispensable in understanding the corrosion phenomena occurring in water-cooled reactors. Therefore, the author had to determine how to modify Pourbaix’s diagrams in consideration of the radiation environment as it is an important research paradigm.
In reactor systems, radiation is an additional energy input onto electrochemical processes. The electrochemistry deals with electric potential energy as well as the energy from chemical reactions (i.e. Gibbs energy). This leads to a new scientific paradigm, of how to integrate radiological and electrochemical energies, which is expressed through the Nernst equation. Although radiation may have an effect on many chemical reactions involved in the corrosion processes, the predominant ones are those from the radiation chemical effects on the reactor water. This is because the concentrations of chemical species involved in corrosion processes (e.g. metallic ions), are several orders of magnitude less than the concentrations of water molecules. The irradiated water being discussed here contains H+ ions, OH− ions as well as stable molecular products through irradiation such as DH, DO and H2O2. Some are dosed for “water chemistry control” in the reactor water.
With the notion that the predominant reactions are those from the radiation effects on the reactor water, it is possible to separate the radiation chemical effects on the reactor water from the electrochemical reactions of metals. This indicates that the E-pH diagrams of metals and compounds can be calculated in a non-radiation environment whose results are applicable to corrosion studies in the irradiated water environment. The chemical- and electrochemical properties of the irradiated water determines the actual corrosion environment under radiation (e.g., ECP and pH). Based on this finding the author has updated his previous electrochemical studies on AOA (Saji, 2015b), as summarized in Annex C.
3.3.2. Effect of radiation on the equilibrium potential of reactor water
Radiation effects on water should result in a change in the “conditional-” or “formal” potential12. Since radiation introduces additional energy on top of the electrochemical energy (i.e., electric potential energy and Gibbs energy of chemical reactions) the Nernst equation is not directly applicable. Although the “standard electrode potentials” of radical species were compiled in 1952 by Latimer (1952), the subsequent edition compiled by IUPAC published in 1985 eliminated this information on radical species (Bard et al., 1985). In this edition, the standard potential is limited to “cell reaction potential against the standard hydrogen electrode.” The radiation chemical reactions are not an electrochemical cell reaction since they occur in bulk water but not at the electrodes. This issue is much more complicated as pointed out in a recent reference (William and William, 2011).
The author sought a way to integrate the additional radiological energy (both electrical potential energy through annihilation of e−aq as well as chemical reaction energy of non-charged radiation-chemical species). Considering the large uncertainties the formal potential was used as a fitting parameter in previous studies. The for the hydrated electron that is estimated by fitting the author’s theoretical curves with the experimentally observed potential difference 0.1/0.3 V for BWR(NWC)/PWR was substantially smaller than the generally accepted standard electrode potential value for of 2.86 V. The difference was greatly reduced by separating the electrochemical portion from the chemical portion of the intermediate reaction steps and by only using the energy that corresponds to the disappearance of the charge induced by the reactions with e−aq. The standard electrode potential value of 2.86 was calculated by assuming a hypothetical intermediate state that includes both electrochemical- and chemical processes in converting the sum into the electrochemical standard potential as summarized in Annex E. Although this value is sometimes quoted in textbooks and handbooks, the experimentally observed potential difference should not be compared with these standard electrode potential values considering these difficulties. Instead, a new convention is introduced which states that the radiation-induced potential difference should converge to zero by decreasing radiation in the following discussion.