1. Introduction
At present, the effective therapy of tumor is still a tremendous challenge for human beings. There are a number of critical drawbacks in traditional chemotherapeutics to overcome, such as harmful side-effects, low therapeutic efficiency, non-specific biodistribution, low circulation time and poor solubility [1], [2]. This can be achieved by encapsulating the therapeutic agents in biocompatible and biodegradable nano-vehicles that can avoid the identification of the immune system and release drugs slowly [3]. Liposomes displayed excellent application prospect for advantageous drug transport, which are a self-assembling structure of a lipid dispersion in water and have been used to encapsulate both hydrophilic therapeutic compounds or lipophilic ones [4]. Liposomes have many advantages, such as increasing drug capacity, versatile structure and facile surface decoration, good biodegradability properties, and targeting and protection of entrapped agents [5], [6]. In addition, as a drug delivery system, nanoparticle size would be a precondition and a crucial factor which decides the fate of drugs both in vivo and in vitro [7]. H. Maeda and co-workers have reported that most solid tumors possess unique pathophysiological characteristics, such as extensive angiogenesis, defective vascular architecture, and impaired lymphatic drainage/recovery system. Thus, tumor vessels were more permeable, so the molecules of certain sizes tend to accumulate in tumor tissue much more than they do in normal tissues. The phenomenon now known as the enhanced permeability and retention (EPR) effect [8]. The size between 100 and 200 nm was acknowledged as the best particle size for EPR effect, which is the main mechanism for passive targeting of nanoparticles to tumor site [9]. Therefore, appropriately sized liposomal carriers can be used to avoid drug extravasation through continuous capillaries of healthy tissues and provide a steric barrier to prevent exposure of the encapsulated drug to healthy cells [3]. So, the drugs with appropriately size distribution are beneficial to accumulate in tumor tissues.
However, targeting drug delivery and controlling drug release at the tumor site are still major challenges for efficient therapy of cancers. The selective and site-specific release drugs have the particular interest for reducing drug toxicity and improving overall therapeutic safety. The surface modification of liposomes has been performed to stabilize the liposomes in normal body environments and avoid the leakage of encapsulated compounds, meanwhile, promote drug rapid release from liposomes at target site [10]. Tumors exhibit a lower extracellular pH than normal tissues, as well as in their intracellular lysosomes and endosomes. Based on this property, a variety of materials have been functionalized to design pH-responsive delivery systems for the site-specific controlled release of payloads by simply exploiting the pH changes [11], [12], [13]. For example, the pH-controlled release of DOX from single-walled carbon nanotubes was successfully applied to in vivo cancer therapy [14]. The pH-responsive NPs of polyethylene glycosylated peptide dendron–doxorubicin conjugates were fabricated for cancer therapy [15]. Chitosan (CS), a natural polysaccharide, is derived from partial deacetylation of chitin of crustacean shell [16]. CS and its derivatives have been employed in many biomedical applications due to the polycation intrinsic properties, low toxicity and excellent biocompatibility [17]. In this study, CS was chosen to modify the liposomes surface, because CS is capable of opening the tight junctions of epithelial cells, resulting in a paracellular pathway through the epithelial barrier [18].
Ursolic acid (UA) is a natural pentacyclic triterpene of the cyclosqualenoid family, which is ubiquitous in the plant kingdom and found in many foods and herbs such as apple, cranberry, rosemary, and oregano [19]. UA, as a kind of bioactive natural compound, possesses a wide range of biological activities including anti-inflammatory, antibacterial, antiviral, anti-diabetes and immunomodulatory activity [1]. However, its most prominent function is anticancer effects [20], as it can influence many different cancer pathways including inhibition of tumor angiogenesis, promotion, proliferation, and metastasis [21]. Kassi et al. successfully used UA to treatment human prostate cancer [22]; UA could completely inhibit the appearance of a palpable tumor in a subset of mice, meanwhile, it would be capable of inhibiting cell proliferation, cell cycle distribution, and apoptosis of human lung cancer cell line A549 [23], [24]. However, the clinical application of UA is limited as its poor water solubility, low bioavailability [25], and short plasma half-life [26]. Therefore, an efficient drug delivery system is desired to overcome the obstacles.
The main aim of the present work was to develop a CS modified drug delivery system to obtain the pH-responsive release and specifically drug accumulation in tumor cells (Scheme 1). The drug delivery system with smaller diameters of ~ 130 nm has been developed by loading of the natural antineoplastic drug UA, and CS was modified on the surface of liposomes. Through the surface modification of CS, the CS-UA-L could selectively target to the tumor site and be taken up by tumor cells. After encapsulating UA in CS-UA-L, its bioavailability could be effectively improved by release of drug slowly at tumor site. Such a pH-responsive delivery vehicle will provide a valuable approach to construct smart and biocompatible agents on tumor treatment.
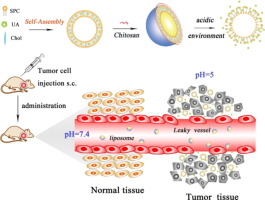
 Scheme 1. Schematic diagram representing the mechanism of synthesis of CS-UA-L and pH-triggered sequential UA release in tumor bearing mouse.
Scheme 1. Schematic diagram representing the mechanism of synthesis of CS-UA-L and pH-triggered sequential UA release in tumor bearing mouse.2. Materials and methods
2.1. Materials and animals
All experimental protocols involving animals were approved by the Animal Subjects Committees at the Yanshan University, which were carried out in accordance to their guidelines and regulations. Ursolic acid was purchased from Hefei Hiromi Biological Technology Co., Ltd. (Shenyang, China); Soybean phosphatidylcholine (SPC) was purchased from Shenyang Tianfeng Biological Pharmaceutical Co., Ltd. (Shenyang, China); Cholesterol (CHOL), anhydrous ethanol and surfactant Tween-80 were purchased from Tianjin Guangfu Fine Chemical Research Institute (Tianjin, China); Chitosan (degree of deacetylation: 92%) was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China), Acetic acid glacial was obtained from Tianjin Fengchuan Chemical Reagent Technologies Co., Ltd. (Tianjin, China). Sephadex G-75® was obtained from Sigma Chemical Company (Henan, China). Hematoxylin and eosin were obtained from Beijing Biodee Biotechnology Co., Ltd. (Beijing, China). All reagents used were of at least analytical grade. HeLa cell was gifted from the first hospital of Qinhuangdao and U14 cervical carcinoma cell line was purchased from the Peking Union Medical College.
CD-1 female mice were purchased from Vital River Laboratory Animal Center (Beijing, China). The mice were maintained under standard conditions of temperature (22–25 °C), a humidity of 50–65% and a 12 h light/dark cycle. The animals were fed with a standard diet of mouse chow, and water was allowed ad libitum. All animal experiments were approved by the Animal Subjects Committees at Yanshan University, and were carried out in accordance to their guidelines and regulations.
2.2. Preparation of UA liposomes and CS-UA-L
UA liposomes (UA-L) were prepared using an ethanol injection method. Briefly, the hydrophobic components SPC, CHOL and UA (in weight ratio of 50: 6: 5) were dissolved in 3 mL of anhydrous ethanol as the lipid phase, stirring gently by the electric heating magnetic stirrer until completely dissolved. Tween-80 (0.1%, v/v) was dissolved in 10 mL phosphate-buffered saline (PBS, pH 6.5) at 43 °C as the aqueous phase. Then, the lipid phase was dropped into the aqueous phase under mild magnetic stirring. The translucent blue opalescent UA-L solution was produced spontaneously after further evaporation of the residual ethanol.
For the preparation of CS-UA-L, 0.1% CS was dissolved in 0.1 M acetic acid glacial solution (pH 3.5) to obtain a CS solution. Then, CS solution was added drop wise to the equal volume of liposomes solution under magnetic stirring at 20 °C for 2 h [27]. Finally, the unencapsulated UA and free CS were removed by gel permeation chromatography through a Sephadex G-75 column (1.0 cm × 25 cm). All the liposome dispersions were stored at 4 °C for further analysis.
The encapsulation efficiencies (EE) of UA formulations were calculated using the following calculations:
EE (%) = weight of drug loaded/total weight of drug added in the preparation × 100%.
The UA amounts in the liposomal formulations were determined by high-performance liquid chromatography (HPLC) (Agilent-1200). The HPLC conditions included a Diamonsil-C18 column (5 μm 4.6 × 250 mm), mobile phase composed of acetonitrile/0.5% acetic acid (90:10), 1 mL/min flow rate, 210 nm detection wavelength, 20 μL injection volume, and 28 °C column temperature.
2.3. Characterization of CS-UA-L
2.3.1. Characterization and stability
The shapes and the structures of the prepared liposomes were investigated by TEM (HT7700, Japan) photos. The negatively stained liposomes were prepared by an upside-down method following a standard procedure. Briefly, 3% tungstophosphoric acid was used as negative staining agents. The negative staining agent and liposome suspension were mixed with 1:3 (v/v), then the carbon film-coated copper grid was placed on the mixed sample for 10 min, and the excess solution was removed with a filter paper. The sample was air-dried under dust-free condition, and then observed by TEM.
AFM (Veeco/Bruker, America) was used to obtain the tridimensional morphology and elastic properties of the liposomes. Just before analysis, 30 μL suspension was dropped onto the surface of a new cleaved mica sheet (about 1 cm), which was air-dried on a super clean workbench for the detection. Freshly cleaved mica was mounted and measured onto a stainless steel disc using a sticky tab (Latham, NY).
To determine CS-UA-L stability in various physiological solutions, the CS-UA-L was added in phosphate buffered saline (PBS) or cell culture medium with 10% fetal bovine serum (FBS) with a volume ratio of 1:1, respectively. Then, the solutions were stored in 4 °C for a period of 5, 10, 15, and 20 days, respectively. At the different time point, the particle size, PDI, zeta-potential and encapsulation efficiency for the liposomes were analyzed. All experiments were performed in triplicate.
2.3.2. Particle size distribution and zeta potential
The hydrodynamic diameters and distribution of the above liposomes were determined by dynamic light scattering (DLS) method using a Malvern Zetasizer Nano ZS (Malvern; Worcestershire, U.K.). Just before analysis, the samples were diluted (20 μg/mL) with deionized water to avoid multiscattering phenomena, and then the same volume of samples were used for analysis. The Zeta potentials of samples were also analyzed using above instrument. All results were the average of triplicate measurements.
2.4. Fourier transform infrared spectroscopy (FTIR)
FTIR spectrums of pure CS, UA-L and CS-UA-L were analyzed in the range of 400–4000 cm− 1 by a FTIR spectrometer. Mannitol and sucrose as lyoprotectant were thoroughly milled with different formulations, then; a vacuum freeze drying machine was used to prepare the lyophilized powder of samples. A small quantity of lyophilized sample was mixed with pre-dried KBr powder, and then the mixture was grind into fine powder by agate mortar. Finally, the FTIR spectra of the samples were recorded on a Nicolet iS10 (USA) spectrometer. Each result was an average of triplicate.
2.5. In vitro drug release study
In order to explore the UA release performance and the pH response of the drug vehicles, drug release effects from CS-UA-L were investigated using the dialysis method in different buffers, which simulated the pH values of tumor (pH 5) and normal physiological environment (pH 7.4) [28], [29]. Briefly, the dialysis bags were pretreated in the usual way, an aliquot of UA-L and CS-UA-L (1 mL) were respectively placed into a dialysis bag with a 12,000–14,000 Da molecular weight and sealed tightly. Then the dialysis bags containing 1 mL liposomes were immersed in 200 mL phosphate buffer (pH 5 or 7.4) containing 0.1% (v/v) Tween 80 to maintain the sink condition (the best condition for drug dissolution). Then, the beakers were placed into water bath at 37 ± 0.5 °C and stirring was maintained at 100 ± 1 rpm. After releasing, the samples (1 mL) were taken from the dialysis bag at predetermined time intervals (2, 4, 6, 8, 10, 24, 36, 48 and 72 h), and the drug content of each sample was analyzed by HPLC. All experiments were performed in triplicate.
2.6. Antitumor effect study in vitro
The inhibiting effects of free UA, UA-L and CS-UA-L for tumor cells were evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoli um bromide (MTT) assay. Briefly, HeLa cells in Dulbecco modified Eagle medium (DMEM) were seeded into 96-well plates at a density of 2 × 104 cells/well, and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. All the samples were filtered through sterile 0.22 μm millipore membrane, and further diluted with sterile PBS (pH 7.4) to adjust UA concentration in the range of 0–250 μg/mL. Each sample was determined in triplicate (100 μL per well) into the appropriate well of culture dishes, and DMEM culture medium was regarded as blank control. After 24 h, the treated-cells were washed twice with PBS (pH 7.4), 100 μL of MTT working solution (0.5 mg/mL) was put into each well and the cells were incubated in a CO2 incubator at 37 °C for 4 h. Medium in wells was removed and replaced with 150 μL of dimethylsulfoxide (DMSO) to dissolve the blue formazan crystals produced by the live cells. The plates were shaken for 5 min before detection, and then the absorbance of sample was obtained by an enzyme-linked immunosorbent assay reader at 490 nm. The tumor cell inhibitory rate was calculated according to the following equation:
Inhibitory rate (%) = (1 − ODsample / ODcontrol) × 100
Where, ODsample and ODcontrol represented the absorbance of the samples and controls, respectively. All experiments were carried out three times.
2.7. Antitumor activity for tumor-bearing mice
Female CD-1 mice (20 g, 6–8 weeks old) were used for pharmacodynamic studies, 200 μL of U14cells (2 × 106) were subcutaneously injected into the right forelimb oxter of CD-1 mice to establish animal tumor model. The solid tumors of mice were palpable after inoculation 4 days, then the mice were randomly assigned to four groups, the detailed groups and treatment methods were as shown in Table 1. Then, 200 μL of saline, free UA solution, UA-L or CS-UA-L were given to the mice via intragastric administration respectively, all the formulations were administrated once a day for a total of 14 days. During the administration, the weight of mice and the sizes of xenografts were measured every 2 days. The tumor sizes were monitored with a vernier caliper, and the tumor volume was calculated as V = 0.5ab2, where a and b are the long and short diameters of the tumor respectively.
Table 1. Groups of animals in experiment.
| Groups | Number | Treatment methods |
|---|---|---|
| 1 free UA (80 mg/kg) | 8 | 5.0 mg/mL UA suspension in twice |
| 2 UA-L (80 mg/kg) | 8 | 5.0 mg/mL UA liposome suspension in twice |
| 3 CS-UA-L (80 mg/kg) | 8 | 5.0 mg/mL UA-CS liposome suspension in twice |
| 4 NC | 8 | Intragastric administration equal volume of normal saline |
NC, negative control; free UA, UA solution; UA-L, UA liposomes; CS-UA-L, chitosan-coated UA liposomes.
Antitumor activities of the samples were assessed with the tumor growth inhibition rate (IR) at the experimental endpoint, which IR % = [(Tumor weight in normal saline control group − Tumor weight in treated group) / (Tumor weight in normal saline control group)] × 100%.
After the final evaluation, the mice were sacrificed by cervical vertebra dislocation, and the tumors were excised and weighed, then the major organs were also excised for further analysis.
2.8. In vivo biodistribution of UA
The CD-1 mice were subcutaneously injected with U14 cells using the above method. When the solid tumors of mice were palpable, the drugs of free UA, or UA-L or CS-UA-L with UA dose of 80 mg/kg body weight were treated via intragastric administration. The mice were sacrificed at 12 h post-administration, and then the interested tissues (tumor, heart, liver, spleen, lung, and kidney) were rapidly removed from each mouse. Each organ was individually homogenized in cool normal saline; the homogenate was transferred to a new tube containing 0.5 mL of acetonitrile, vortexed to precipitate protein, and then centrifuged at 10,000 rpm for 20 min. Finally, the supernatant was filtered through a 0.22 μm membrane filter and analyzed for UA content via HPLC.
2.9. Histopathological study
All mice were sacrificed on the fifteenth day, and the tumor tissues were carefully removed and fixed in 4% paraformaldehyde solution for 24 h at the room temperature. Then the tissues were embedded in paraffin wax and the sections of 3–5 μm thickness were obtained using a rotatory microtome (Leica Company, Germany), and stained with Hematoxylin and eosin (H&E). The microstructure and morphology of tissues were observed by a light microscopy.
2.10. Statistical analysis
Descriptive statistics included the mean and standard error. Statistical analysis was conducted using analyses of variance (ONE WAY ANOVA), with P < 0.05 considered to be statistically significant.
3. Results and discussion
3.1. Characterization of liposomes
3.1.1. Characterization
Morphological characterization of the liposome formulations was performed by TEM and AFM, whose structures were shown in Fig. 1, which displayed spherical morphology. The liposomal particle size with CS modification was slightly increased compared to the non-modified liposomes, which because CS formed a hydrophilic shell on the liposomes surface. The interaction between CS and liposomes is due to the electrostatic interactions between positively charged CS and negatively charged phosphates [30]. In addition, when the CS coated on the surface of the liposomes, the CS layer can make liposomes loading much positive charges, and the charged liposomes are benefited on the stability of carriers. The stability of liposomes can be increased due to the fact that the same charges produce electrostatic repulsion, which will cause reject among liposomes and avoid aggregation and fusion.
 Fig. 1. TEM and AFM images of liposomes. (A), (B) conventional UA liposomes; (C), (D) chitosan coated UA liposomes.
Fig. 1. TEM and AFM images of liposomes. (A), (B) conventional UA liposomes; (C), (D) chitosan coated UA liposomes.AFM was used to further explore the stereostructure of the liposomes, the observed particles were regular spherical with narrow size distribution and good dispersivity. The heights of the UA-L and CS-UA-L were about 55.2 nm and 166.5 nm, respectively. The ratio of the height to the particle diameter reflects the rigidity of liposomal vesicles and the surface deformation degree of liposomes on mica [31]. As shown in Fig. 1, the CS-UA-L presented a hemisphere without intermediate concave; meanwhile, surface deformation and middle sag were appeared on the non-modified liposomes. Thus, the rigidity of CS-UA-L was stronger than that of the ordinary liposomes. Several researchers have been reported that the rigidity of the carrier particle is one of the most important properties affecting drug delivery effectiveness, which can affect the particle stability, release profile of encapsulated drug, and blood circulation time [31].
3.1.2. Particle size and zeta potential
Fig. 2 displayed the particle sizes of the different liposomes prepared. The average diameter of UA-L determined by DLS was 120.4 ± 0.473 nm (Fig. 2A), and the mean size of CS-UA-L was 135.4 ± 0.636 nm, which attributed to CS formed a hydrophilic shell on the liposome surface via anchoring the hydrophobic groups on the phospholipid bilayer of liposomes. The polydispersity indexes (PDI) for all the formulations were smaller than 0.3 (Fig. 2), which indicates a relative homogeneous dispersion [32]. In addition, the research found that the size between 100 and 200 nm was acknowledged as the best particle size for EPR effect, which is the main mechanism for passive targeting of nanoparticles to tumor site [9]. Therefore, the CS-UA-L with the mean diameter of 135 nm was more conducive to targeting the tumor tissues.
 Fig. 2. Mean Particle Sizes, Polydispersity Indexes (PDI) of (A) UA-L and (B) CS-UA-L. (C) Zeta potentials of the UA-L and CS-UA-L.
Fig. 2. Mean Particle Sizes, Polydispersity Indexes (PDI) of (A) UA-L and (B) CS-UA-L. (C) Zeta potentials of the UA-L and CS-UA-L.Zeta potential has often been used for characterizing colloidal drug delivery systems, which is a detection index of the surface electrical charge of the particles. In addition, the surface potential is an important parameter influencing liposomal behavior and function. In vivo, the surface charge density has been found to influence the distribution of liposomes; and in vitro, a high potential might contribute to the physical stability of liposomes by reducing the rate of aggregation and fusion [16]. Fig. 2C showed that the CS modification changed the zeta potential of UA-L, which was elevated from slightly negative value (− 6.7 mV) to positive value (7.8 mV). As a cationic polymer, the adsorption of CS on the surface of the liposomes increased the density of the positive charges and elevated the zeta potential of the drug carrier [33].
3.1.3. Stability studies
To determine the stability of liposomes in various physiological solutions, CS-UA-L was incubated respectively in PBS or cell culture medium with 10% FBS at 4 °C for 20 days. No apparent change was observed on the particle size, PDI and zeta-potential of CS-UA-L (Table 2). Besides, the encapsulation efficiencies of CS-UA-L decreased lightly after stored in PBS for 20 days. The same phenomenon was found after the samples were stored in cell culture medium with 10% FBS (Table 3). All these results indicated that CS-UA-L had an excellent stability. In addition, the UA-CS-L exhibited excellent dispersity in PBS or cell culture medium (Fig. 3), and no aggregate was found by the naked eye or optical microscopy.
Table 2. Sizes, PDI, zeta-potentials and encapsulation efficiencies (EE) of CS-UA-L after storage in PBS.
| Parameters | Time (days) | |||
|---|---|---|---|---|
| 5 | 10 | 15 | 20 | |
| Size | 135.1 ± 1.61 | 141.7 ± 1.48 | 146.4 ± 1.08 | 151.7 ± 0.96 |
| PDI | 0.22 ± 0.012 | 0.2 ± 0.006 | 0.22 ± 0.012 | 0.19 ± 0.084 |
| Zeta | 6.55 ± 0.41 | 8.68 ± 1.43 | 9.18 ± 0.48 | 11.9 ± 0.82 |
| EE | 91.4 ± 4.49 | 87.7 ± 4.33 | 85.5 ± 2.73 | 83.85 ± 2.77 |
All values were expressed as mean ± S.E. (n = 3).
Table 3. Sizes, PDI, zeta-potentials and encapsulation efficiencies (EE) of CS-UA-L after storage in serum medium.
| Parameters | Time (days) | |||
|---|---|---|---|---|
| 5 | 10 | 15 | 20 | |
| Size | 135.4 ± 0.64 | 141 ± 0.75 | 144.6 ± 2.04 | 147.4 ± 1.15 |
| PDI | 0.21 ± 0.005 | 0.2 ± 0.058 | 0.22 ± 0.015 | 0.19 ± 0.108 |
| Zeta | 6.06 ± 0.14 | 7.64 ± 0.98 | 9.55 ± 1.64 | 10.56 ± 2.45 |
| EE | 90.9 ± 3.3 | 88.6 ± 3.19 | 87.1 ± 3.91 | 84.1 ± 6.3 |
All values were expressed as mean ± S.E. (n = 3).
 Fig. 3. Digital images of CS-UA-L after incubation with PBS (left) or DMEM with 10% FBS (right) for 3 days.
Fig. 3. Digital images of CS-UA-L after incubation with PBS (left) or DMEM with 10% FBS (right) for 3 days.