1. Introduction
The world-wide plastic crisis is real. With 4.8 billion tons of plastic in poorly managed landfills (Geyer et al., 2017), with close to 400 Mt of new plastic produced in 2020 (Hundertmark et al., 2018a), and with less than 10% of this new plastic recycled even once (less than 1% recycled twice (Geyer et al., 2017)), we face a daunting challenge. While plastic in the environment reached the public debate in many countries, the outlook is dim: Borelle et al. tell us that even in a react-now scenario (“ambitious”), 20 to 50 Mt of plastic will be disposed into aquatic ecosystems every year by 2030 (Borrelle et al., 2020). Another challenge of the ever-increasing plastic use, reaching 1000 Mt by 2050 (Hundertmark et al., 2018a), is the use of fossil resources. Indeed, the chemical industry is estimated to have the highest growth rates for fossil resources by 2030 (International Energy Agency, 2018). The resulting annual greenhouse gas emissions would reach 6.5 Gt CO2 equivalents by 2050 (Zheng and Suh, 2019). Surely, the overall challenge cannot be changed by the dismal contribution of clearly less than half a percent of plastic produced from renewable carbon sources, i.e., biomass, CO2 (plus green hydrogen in the future (Blank et al., 2020)), and waste streams yet. However, sugar as carbon source is gaining momentum, e.g., for polylactic acid (PLA) produced from lactic acid (LA). A new PLA production plant is announced by TotalCorbion to be built in Europe, with a yearly production capacity of 125,000 t PLA, and other developments around the world, especially in China. The production capacity of microbial polyesters (poly-3-hydroxybutyrate (PHB) and polyhydroxyalkanoates (PHA) of various monomer compositions) are in the 5000 tonnes per annum range (e.g., Kaneka, Danimer Scientific). While it is exciting to see that bioplastic finally excels, the absolute contributions to the plastic market are currently very small.
Emergence of a sustainable plastics economy will strongly rely on fair pricing, e.g., of CO2 including its climate impact, as implemented for other industrial sectors in the EU Emissions Trading System. Wei et al. argued in a recent commentary for a zero fossil resource plastic economy, relying on the “6 R” principles - rethink, refuse, reduce, reuse, recycle, and replace (Wei et al., 2020). Such a future plastic economy would partially rely on biotechnological technologies for production and end-of-life plastic treatment (Lee et al., 2011). Importantly, all plastic that might end up in the environment should be equipped with an emergency degradation mechanism, which overcomes the observed accumulation of plastic waste in the environment. Rubber, in form of tiny particles from tire wear might be such an example, as it is estimated that in Germany alone, 100,000 tonnes per annum are lost into the environment, while no major accumulation/sinks are known to date. Indeed, a half-life of about 16 months was reported (Cadle and Williams, 1980) and vulcanized rubber is known to be biodegradable (Rose and Steinbüchel, 2005), indicating such an emergency degradation. While atmospheric oxidation seems the main mechanism, two evolutionary different enzyme systems are known that can attack the double bond in rubber (Yikmis and Steinbüchel, 2012; Jendrossek and Birke, 2019; Birke et al., 2018). Reliable studies that quantify the fate of tire wear in the environment are however urgently needed.
While environmental plastic degradation is discussed for decades and examined for example by ISO norms ISO/DIS 23832 and ISO 14855–2, the degradation to oligomers and monomers for use as substrate for microbes has received less attention. In 2018, the European Union established the “European Strategy for Plastics in a Circular Economy”, funding plastic-based biotech. In this framework, the MIX-UP project focuses on changing the traditional linear value chain of plastics to a sustainable, biodegradable based one (Ballerstedt et al., 2021). Here, we briefly summarize the state-of-the-art of two alternative technologies to obtain oligomers and monomers: 1. Enzymatic plastic degradation and 2. Plastic degradation by pyrolysis. We are aware of the intensive efforts on chemical plastic recycling (Ellis et al., 2021; Vollmer et al., 2020; Weckhuysen, 2020), with exciting examples like combined polymer degradation and monomer hydrogenation (hydrogenolysis) of polyethylene terephthalate (PET) and PLA to the corresponding diols (Westhues et al., 2018). The chemical advance in PET recycling culminated in rapid hydrolysis at room temperature, a catalytic challenge that was not expected to be solved so rapidly or at all (Tanaka et al., 2021). In addition, some modified PE-like polymers were suggested, with performance parameters much resembling PE, however equipped with activatable bonds for novel end-of-life options (Baur et al., 2021; Häußler et al., 2021). Chemical plastic recycling advances are, however, not in the scope of this review.
Here, we evaluate the potential of plastic monomers as microbial substrates. With the molecular constituents of potential plastic hydrolysates in hand, we summarize their catabolic pathways and compute the theoretical yields for the production of common or potential biotechnology products (Fig. 1). Finally, we use these theoretical biochemical yields to compute replacement scenarios of products currently produced from fossil resources. The results might guide future metabolic engineering efforts, in which plastic waste is used for upcycling to generate (plastic) value. In addition, the results also highlight the shortcoming of some of the investigated native biochemical pathways and argue for synthetic pathways that support chemical synthesis routes with attractive substrate-to-product yields.
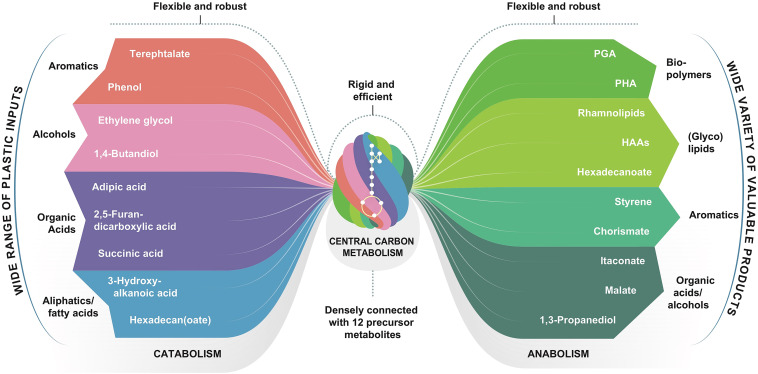
 Fig. 1. The bow tie of microbial metabolism. The flexibility of the microbial metabolic arsenal facilitates the conversion of plastic monomers to valuable products. While the central carbon metabolism is rigid, the catabolic (left) and anabolic (right) pathways show a high versatility and are amenable to metabolic engineering. This versality allows researchers to modify microorganisms for the usage of many different monomers from post-consumer plastic waste for the production of a plethora of industrially interesting compounds. The groups of substances on the substrate side include monomers from industrially relevant polymers and represent a selection (e.g., amines are missing). The products are industrially interesting molecules, already synthesized or suggested to be synthesized using microbes. PGA – polyglutamic acid, PHA – polyhydroxyalkanoates, HAA – hydroxyalkanoyloxy alkanoate.
Fig. 1. The bow tie of microbial metabolism. The flexibility of the microbial metabolic arsenal facilitates the conversion of plastic monomers to valuable products. While the central carbon metabolism is rigid, the catabolic (left) and anabolic (right) pathways show a high versatility and are amenable to metabolic engineering. This versality allows researchers to modify microorganisms for the usage of many different monomers from post-consumer plastic waste for the production of a plethora of industrially interesting compounds. The groups of substances on the substrate side include monomers from industrially relevant polymers and represent a selection (e.g., amines are missing). The products are industrially interesting molecules, already synthesized or suggested to be synthesized using microbes. PGA – polyglutamic acid, PHA – polyhydroxyalkanoates, HAA – hydroxyalkanoyloxy alkanoate.2. Polymers to monomers
In recent years, biotechnological approaches have been proposed as sustainable alternative for plastic recycling (Wei et al., 2020). The simplest mechanism for depolymerizing plastics is the direct microbial depolymerization of the polymer, which can also occur in environments contaminated with plastic. For more recalcitrant polymers like PET, enzymatic depolymerization requires dedicated enzyme reactors under specific working conditions like increased temperatures. Polymers containing highly recalcitrant C–C bonds in their backbones for which no dedicated enzymes are reported (yet), might be degraded over time by unspecific oxidases (e.g., laccases, peroxidases), however at very low rates or not at all depending on the environmental conditions. These plastics can be depolymerized technically by, e.g., pyrolysis, obtaining a condensate, the pyrolysis oil. In the following section, these different methods are described, starting with microbial degradation and progressing viaenzymatic cleavage to pyrolytic plastics degradation.
2.1. Microbial depolymerization
Microbial depolymerization usually does not yield monomers for subsequent processing, since microbes take up the monomers directly and generate biomass and CO2. We will in this section only focus on the depolymerization and the formation of the respective monomers (Table 1).
Table 1. Monomers from microbial deconstruction of biodegradable polymers.
| Polymer | Monomers | Refs |
|---|---|---|
| Polyhydroxyalkanoates | 3-hydroxyalkanoic acid | Chowdhury (1963) |
| Polybutylene succinate | 1,4-butanediol, succinic acid | Tokiwa et al. (2009) |
| Polybutylene adipate terephthalate | 1,4-butanediol, adipic acid, terephthalic acid | Utomo et al. (2020) |
| Polylactic acid | D-lactic acid, L-lactic acid | Zaaba and Jaafar (2020) |
Polymer utilization by microbes is so common that we usually do not give it any attention, especially for sugar polymers like cellulose or aromatic polymers like lignin. While these processes occur in nature, the technical realization of the lignocellulosic polymer degradation and subsequent monomer utilization still waits for its economic breakthrough, despite huge efforts in the last decades. This is in no small part due to the recalcitrance and complexity of lignocellulosic biomass as biotechnological substrate, requiring harsh pretreatment and complex enzyme cocktails to be degraded at biotechnologically relevant rates.
In contrast, starch is effectively used as biotechnological feedstock at very large scale due to its ease of purification and simpler structure (Mussatto et al., 2010). These features enable biotechnological utilization of plastics as carbon source, since purity and bioavailability of starch are similar to biodegradable polymerssuch as PHA, polybutylene succinate (PBS), polybutylene adipate terephthalate (PBAT), and polylactic acid (PLA). Starch is also widely used as biodegradable polymer for packaging and consumer goods (nova-Institute, 2020). Microbes able to degrade and assimilate polymers traditionally considered non-biodegradable were rarely found (Ru et al., 2020), although also in this field much development is ongoing. One example is the already iconic Ideonella sakaiensis isolate, which can depolymerize amorphous PET and grow partly on the released monomers, albeit very slowly (Yoshida et al., 2016). While the literature is full with reports on microbial plastic degradation including recalcitrant carbon-carbon bond containing plastics, the details are rarely encouraging. A recent study from Kim et al. (2021) suggested Acinetobacter and Pseudomonas strains that are capable of polystyrene degradation.
2.1.1. Polyhydroxyalkanoates (PHA)
PHAs are polyesters with side chains of different lengths: A short chain length PHA is for example PHB, while medium chain length PHAs feature side chains with six to fourteen carbon atoms. PHA polymers are thermoplastic and can be processed on conventional processing equipment (Cataldi et al., 2020), with potential applications in various fields such as biomedicine including tissue engineering, bio-implants, and drug delivery (Raza et al., 2018). Despite decades of research, the total global production capacity of PHAs is sub 50,000 tonnes per annum. That said, a rapid increase is envisaged due to technology readiness, CO2 neutrality, and demand for degradable plastics. Since PHAs are internal carbon storage polymers in many microorganisms, the ability to metabolize PHAs is essential for PHA producers. However, post-consumer PHA is obviously available only extracellularly. The ability to degrade extracellular PHAs depends on extracellular carboxylesterases called PHA depolymerases (Jendrossek and Handrick, 2002; Lee and Choi, 1999), which not all PHA producers feature. While the commonly used PHA producer Pseudomonas putida KT2440 for example does not have an extracellular PHA depolymerase (de Eugenio et al., 2007), microbes such as the bacterial predator Bdellovibrio bacteriovorus possess such an enzyme (Martinez et al., 2012). The monomer resulting from PHA depolymerization is the respective hydroxyalkanoic acid, a potential substrate of β-oxidation in many microbes (see also below).
2.1.2. Polybutylene succinate (PBS)
The applications of PBS overlap partially with polypropylene, however usually with less favorable physical properties. For PBS synthesis, an excess of 1,4-butanediol (1,4-BDO) is used in the first step to obtain BS oligomers. These oligomers are transesterified to high molecular weight PBS under vacuum and in the presence of a chemical catalyst. The world market of PBS is about 80,000 tonnes per annum (nova-Institute, 2020), while substantial investments have been announced in the first two months of 2021 in China alone, adding an additional 900,000 tonnes per annum in the coming years (news from Hengli Petrochemical Co., Ltd.). This rapid development is due to policy changes in China, specifically from “plastic restriction order” to “plastic prohibition order”, which force products made of non-degradable plastic like current bags and disposable plastic tableware gradually out of the market, leading to rapid growth of the same products from degradable plastic. Huge metabolic engineering efforts resulted in market-ready production of 1,4-BDO (Burgard et al., 2016) and succinic acid (SA) (Chae et al., 2020) from renewable carbon sources. Still, most PBS monomers are synthesized by chemocatalysis from fossil resources.
PBS is considered biodegradable (Tokiwa et al., 2009) and this degradation can be rapid. Under laboratory conditions, a thermophilic Microbispora rosea strain degraded 50% of the PBS in eight days (Jarerat and Tokiwa, 2001). As described for other polyesters, esterases and cutinases show activity on PBS, while this activity depends heavily on polymer accessibility and the other physical conditions during hydrolysis. The resulting monomers 1,4-BDO and especially succinate are readily used as carbon source by many microbes (see monomer description below).
2.1.3. Polybutylene adipate terephthalate (PBAT)
PBAT is used in niche applications, in which biodegradation is practical, e.g., in mulching films, often in a mix with plastics like PLA. PBAT is synthesized in two reactions from the monomers 1,4-BDO, adipic acid (AA), and terephthalic acid (TPA). The two intermediates 1,4-BDO/AA polymer and 1,4-BDO/TPA polymer are transesterified to the corresponding co-polymer (Jian et al., 2020). This random co-polymerization explains also why PBAT has no crystalline nature and is therefore accessible for enzymatic degradation. The world market was about 280,000 t in 2020 (nova-Institute, 2020), however, in the recent months, construction of an additional 600,000 tonnes per annum capacity was announced, again mainly driven by policy changes in China. While 1,4-BDO and AA can be biotechnologically produced, chemocatalysis from fossil resources still dominates.
The degradation of PBAT has been reported by enzymes like esterases (Wallace et al., 2017) and cutinases (Soulenthone et al., 2021), but also by single microbes (Soulenthone et al., 2021) and aerobic (Meyer-Cifuentes et al., 2020) or anaerobic (Perz et al., 2016) microbial consortia. Indeed, PBAT is hydrolyzed under favorable conditions like industrial composting in 60 days or even faster (Müller et al., 2001). PBAT-PLA mulching films can be left by the farmer on the field also in moderate climate, as biodegradation takes place under non-favorable conditions, although at a considerably lower rate (Kijchavengkul et al., 2008). The monomers can be utilized by microbes as sole carbon sources (Utomo et al., 2020) (see below for details).
2.1.4. Polylactic acid (PLA)
The basic mechanical properties of PLA are between those of polystyrene (PS) and PET (Lunt, 1998). While PLA can be directly synthesized by microbes (Jung et al., 2010a), the main production route is via chemical polymerization of microbially produced monomers. About 90% of the world-wide produced LA is derived from microbial fermentations. The production of PLA is estimated to be at least 800,000 tonnes per annum by 2020 with Japan and the USA being the two major producers (Karamanlioglu et al., 2017). The construction of 125,000+ tonnes per annum installations are announced in France and China. Being an aliphatic polyester, PLA is in principle susceptible to biodegradation, and this biodegradability and other properties can be tuned by addition of glycolic acidas co-monomer (Jem and Tan, 2020). In recent years, a few thousand studies on enzymatic and microbial degradation have been published (Zaaba and Jaafar, 2020). Several microbial species are able to degrade PLA. Usually, degradation is initiated by excreted depolymerases and subsequent uptake and metabolization of oligomers, dimers, and monomers (Zaaba and Jaafar, 2020). However, in comparison to other biodegradable materials, PLA is more recalcitrant, which results in inefficient microbial degradation (Qi et al., 2017). Enzymatic degradation of PLA is catalyzed by lipases, esterases, and proteases such as alcalases resulting in the release of LA (Zaaba and Jaafar, 2020). While degradation of PLA in natural conditions (e.g., in soil) is very slow, composting under industrial conditions is significantly faster (Ho et al., 1999).
2.2. Enzymatic plastic degradation
As mentioned before, microbes able to degrade and assimilate synthetic polymers were rarely found and furthermore feature very low degradation rates. The history of plastic waste in the environment is with less than 100 years short. Natural evolution for the emergence of degradation mechanisms happens on significantly longer timespans. Thus, for polymers with very low bioavailability the adaption of the microbial metabolic clusters appears to be not fully triggered (Bornscheuer, 2016; Krueger et al., 2015). Consequently, identified microbial enzymes with notable depolymerization activities are likely still the result of moonlighting activities, mainly limited to those active on polyesters or other heteroatomic polymers with ester bonds (for example, polyester polyurethanes (PUR)) (Wei and Zimmermann, 2017a; Danso et al., 2018; Jönsson et al., 2021). Monomers released during enzymatic plastic degradation are diverse (Table 2).
Table 2. Possible monomers arising from enzymatic depolymerization.
| Polymer | Monomers | Refs |
|---|---|---|
| Polyethylene terephthalate | Ethylene glycol, terephthalic acid | Wei & Zimmermann, 2017b |
| Polyethylene furanoate | Ethylene glycol, 2,5-furandicarboxylic acid | Austin et al., 2018; Weinberger et al., 2017 |
| Polyurethanes | Adipic acid, 2,4-toluenediamine, 4,4′-methylene dianiline, 1,4-butanediol, diethylene glycol, 6-hydroxyhexanoic acid | Magnin et al., 2019b, 2020 |
| Polyamides | Adipic acid, azelaic acid, sebacic acid, suberic acid, hexamethylenediamine, 6-aminohexanoate | Kinoshita et al., 1981; Kakudo et al., 1993 |
2.2.1. Polyethylene terephthalate (PET)
As the most widely used polymer type in beverage bottles and synthetic fibers, PET can be synthesized by condensation reactions that start with the monomeric compounds TPA and ethylene glycol (EG). Both monomers can be readily produced by a hydrolytic reaction of amorphous PET using various enzymes (Wei and Zimmermann, 2017b). The first reported polyester hydrolasewith considerable depolymerization activity on bulky PET was a cutinase (TfH) identified in the thermophilic actinomycete Thermobifida fusca (Müller et al., 2005). Using 0.5 mg purified TfH, up to 54% weight loss from approximately 20 mg melt-pressed PET bottle waste (10% crystallinity) was achieved within 3 weeks of incubation at 55 °C. This corresponds to a depolymerization rate of 0.043 mgPET h−1 mgenzyme−1 by converting PET polymer into TPA and EG at an enzyme concentration of 25 mg per gram of PET (mgenzyme gPET−1). Further studies gradually increased this conversion rate to 9.3 mgPET h−1 mgenzyme−1using homolgous T. fusca cutinases (Wei et al., 2014) based on either engineered process with ultrafiltration (Barth et al., 2015) or engineered enzyme variants with improved activities (Wei et al., 2016; Furukawa et al., 2019). More recently, thermostable T. fusca cutinase expressed in Bacillus subtilis has depolmyerized low-crystalline post-consumer PET packaging at 70 °C at a rate of >3 mgPET h−1mgenzyme−1 (Wei et al., 2019a). A higher reaction temperature up to 75 °C was found to be favorable for PET degradation as a result of increased polymer chain mobility (Falkenstein et al., 2020; Wei et al., 2019b). This is also evident because, for example the I. sakaiensis PET hydrolase (IsPETase) with an optimal reaction temperature at 40 °C showed at least two orders of magnitude lower specific activities in degrading the Gf PET films than its thermophilic counterparts active at 65 °C (Tournier et al., 2020). Among these thermostable and -active PET hydrolases, the leaf compost metagenome-derived cutinase LCC (Sulaiman et al., 2012) was found to be the superior biocatalyst as evidenced by the outstanding stability and activity against various amorphous PET materials of the wild-type enzyme at 70 °C (Falkenstein et al., 2020; Wei et al., 2019b; Sulaiman et al., 2014; Tiso et al., 2021). By semi-rational protein engineering, the powerful LCC variant ICCG has been created recently and revealed a maximum depolymerization rate of above 120 mgPET h−1 mgenzyme−1 against amorphized post-consumer PET bottles, corresponding to a maximum productivity of 42 g of TPA per liter per hour (Tournier et al., 2020). This rate was equivalent to a >2800-fold improvement compared to that with TfH against similarly pretreated PET waste as reported in 2005. Moreover, the TPA was readily recovered and purified and finally proven to deliver a comparable product quality in the synthesis of virgin PET as those obtained with petroleum-derived TPA, thereby closing the recycling loop. Alternatively, the PET hydrolysates obtained with LCC have been used as carbon source for engineered P. putida to produce other value-added chemicals by enabling the one-pot use of both TPA and EG, also providing options for open-loop upcycling (Tiso et al., 2021; Meys et al., 2020).
2.2.2. Polyethylene furanoate (PEF)
During the last decade, PEF, a polyester synthesized from 2,5-furandicarboxylic acid (FDCA), has received attention from both scientific communities and industry as a bio-based alternative to PET, both for the fossil fuel saving purpose, and for superior polymer properties (Eerhart et al., 2012). As a result of the reduced flexibility of the furan moieties in PEF, a higher glass transition temperature and corresponding lower chain mobility has been determined in PEF compared to PET (Burgess et al., 2014). Nevertheless, using both the mesophilic IsPETase variants and thermophilic PET hydrolyzing cutinases, enzymatic hydrolysis of PEF has yielded comparable or even higher amounts of the degradation product FDCA compared to those determined with specifically synthesized PET samples in a similar manner (Austin et al., 2018; Weinberger et al., 2017). These results confirmed the broad applicability of various PET hydrolases also for a future scenario. Due to the lack of market-mature PEF products, a biotechnological process for recovered monomers from PEF is, however, still subject of ongoing research, although much can be learned from works focused on the biotechnological production of FDCA in this respect (Saikia et al., 2021).
2.2.3. Polyurethanes (PUR)
Due to the more recalcitrant bonds in PUR (e.g., carbamate, amide and ether bonds), these polymers have so far not been completely depolymerized enzymatically. To date, solid experimental studies overwhelmingly report enzymes cleaving the ester bonds in polyester PUR. Recent reviews summarize the latest findings, including the use of ureases and amidases/proteases for PUR depolymerization (Magnin et al., 2020; Skleničková et al., 2020; Kemona and Piotrowska, 2020; Liu et al., 2021).
The versatility of PET-hydrolyzing cutinases has for example been verified for the degradation of polyester PUR as well as for other petrochemical polymers containing ester bonds (Schmidt et al., 2017; Bollinger et al., 2020). With the superior enzyme LCC, 3.2% weight loss of a commercial thermoplastic polyester PUR was determined following 100 h degradation at 70 °C. This corresponds to a degradation rate of approximately 0.5 mgPUR h−1 mgenzyme−1, which is at least one order of magnitude lower than those with PET described above. The degradation of the urethane bond was reported in a recent patent (EP3587570A1), while the amide bond could be partially degraded using enzymes from fungi (Magnin et al., 2019a, 2020). However, these enzymatic activities were so far still verified with small-molecule PUR model compounds rather than bulk polymers. Various analytic approaches have been applied in the identification of PUR degradation products (Magnin et al., 2019b). Nonetheless, due to the highly variable chemical compositions and polymer structures of commercial PUR products which confine their biodegradability, processes aiming at the recovery and purification of specific PUR monomers derived from biocatalytic upcycling have not been yet well established.
2.2.4. Polyamides (PA)
PA can consist of various aliphatic, semi aromatic, and aromatic units leading to a variety of natural and synthetic polymer types. Among the latter, aliphatic PA such as PA 6 (polycaprolactam, Nylon-6), PA 6.6 (polyhexamethylene adipamide) or PA 6.12 (polyhexamethylene dodecanediamide) are most prominent and mainly used in the textile and automotive industry (Palmer, 2001). Due to the vast number of PA types, many different monomers might be released by enzymatic degradation that could be used as substrates for microbial growth. Depending on the PA, three groups of monomers are expected as degradation products: dicarboxylic acids (such as AA), diamines (such as hexamethylenediamine) and amino acids (e.g., 6-aminohexanoate). Although amide bonds are common in nature, the biodegradation of high molecular weight PA is very rare. To our current knowledge, a manganese-dependent peroxidase (MnP) that was purified from a white rot fungus is the only enzyme reported to degrade high molecular weight PA fibers and membranes (Deguchi et al., 1998). Interestingly, the reaction mechanism for PA degradation partly differs from that reported for lignin-degrading MnPs. Nonetheless, enzymatic degradation of PA by MnP probably occurs in an unspecific way resulting in occasional chain-scission rather than depolymerization of the polymer.
In contrast, three so-called nylonases were identified in Arthrobacter sp. KI72, isolated from sludge near a PA 6 manufacturing plant that specifically degraded cyclic and linear 6-aminohexanoate (Ahx) oligomers. NylA (KEGG, EC 3.5.2.12) was found to convert a cyclic dimer into a linear dimer, showing no activity towards longer cyclic or linear oligomers (Kinoshita et al., 1977; Yasuhira et al., 2010). Ahx linear oligomers were hydrolyzed by NylB (KEGG, EC 3.5.1.46) viaan exo-type hydrolysis or by the endo-acting NylC (KEGG, EC 3.5.1.117) (Kinoshita et al., 1981; Negoro et al., 1992). NylB showed highest activity towards the linear dimer, generating Ahx. The activity decreased with increasing chain-length of linear oligomers resulting in a relative activity of 8% on the hexamer and 0.3% on the icosamer compared with the linear dimer (Chibata et al., 1982). NylC degraded Ahx linear oligomers with a degree of polymerization >3 with a preference for the pentamer, and also showed activity towards the Ahx cyclic tetramer (Kakudo et al., 1993, 1995). Enzyme engineering has led to a thermo-stabilized NylC that was able to degrade thin-layered PA 6, PA 6.6, and PA 6.6-co-6.4 in which succinyl units were incorporated (Negoro et al., 2012; Nagai et al., 2014). Hence, nylonases are promising candidates for further enzyme engineering approaches to design highly efficient PA depolymerases.
2.3. Pyrolytic plastic degradation
Purely biotechnological approaches are not suited for depolymerizing polymers more recalcitrant than the ones described so far (e.g., polyethylenes and polypropylenes with their stable carbon-carbon bonds). In this case, interdisciplinary approaches are required that combine highly efficient thermochemical or catalytic depolymerization methods with microbial metabolization. One example for such an efficient depolymerization method is pyrolysis.
Pyrolysis describes the endothermic process of thermochemical decomposition of material in the absence of oxygen or in an atmosphere of inert gases (Buekens and Meyers, 2012; Panda et al., 2010). Due to increased temperature and heat applied, drying, evaporation, degassing, and chemical cracking processes occur consecutively (Scholz et al., 2001). The decomposition process is characterized by a high complexity of the occurring reactions and mechanisms, which are discussed to depend mainly on the molecular structure of the raw materials. The proportion of solid, condensable, and gaseous products depends strongly on operating parameters such as temperature, heating rate, residence time (gas and solid), and reactor design (Buekens and Meyers, 2012; Quicker, 2020).
Degrading organic macromolecules like plastics into smaller molecules or oligomers and monomers, requires temperatures above 350 °C. Polyolefins, such as polyethylene (PE) and polypropylene (PP) attain a maximum rate of decomposition at ca. 450 °C. Higher temperatures generate on average shorter chain fragments, as cracking is targeted at chain ends first, and then successively proceeds along the polymeric length (Buekens and Meyers, 2012; Panda et al., 2010).
The derived solid residue contains inorganic components (e.g., mineral impurities, fillers from plastics) and carbonized char of high calorific value and carbon content (Czajczyńska et al., 2017). Heavy metals like Cd, Pb, Zn, and Cu concentrate in the solid residues but may also be released partially into the volatile phase at high temperatures and rapid heating rates (Quicker, 2020; Yu et al., 2016). Pyrolysis may also be called ‘dry distillation’ as the yielded pyrolytic gas can be divided into gaseous products and a condensable fraction of liquid or wax consistency at ambient temperature. In general, the condensate comprises paraffins, olefins, and BTX (benzene, toluene, xylene) aromatic compounds. The presence of oxygen leads to the formation of decomposition water, methanol or formaldehyde (Scholz et al., 2001; Ragaert et al., 2017). The organic phase of the condensate is commonly known as ‘pyrolysis oil’ and can be further processed into products by distillation and refining steps. The gaseous products consist mainly of permanent gases and a variety of olefin gases (Scholz et al., 2001). Some polymers contain halogens or other possible pollutants that improve the material properties when integrated into the macromolecule. The degradation of polyvinyl chloride (PVC) for example leads to the formation of gaseous hydrochloric acid. Integrated sulphur compounds of other polymers may enter the gas phase in low quantities as hydrogen sulphideor carbonyl sulphide when degraded (Quicker, 2020).
Highly fluctuating product distribution and low reproducibility pose a great challenge to process control for industrial application of pyrolysis if mixed plastic waste streams are processed. Additionally, certain impurities may have a catalytic effect or can inhibit reactions and substantially affect the product distribution and composition (Ragaert et al., 2017). However, pyrolysis of mono-fraction waste streams such as polystyrene (PS), PA, and polymethyl methacrylate (PMMA) can be adapted by optimizing heating and cooling rates to yield products containing mostly their respective monomers recovering high value chemical precursors (Czajczyńska et al., 2017; Lehrle et al., 2000). PA formed by ring-opening can be transformed into its respective monomers for purification and repolymerization, for example, PA 6 to caprolactam (Punkkinen et al., 2017). PE and PP show random chain fragmentation along polymer length resulting in a broad naphtha-like product spectrum characterized by branched chain products (Table 3) (Czajczyńska et al., 2017; Ragaert et al., 2017).
Table 3. Possible compounds of pyrolysis condensate by thermal degradation.
| Plastic polymer | Compounds of pyrolysis condensate | Refs |
|---|---|---|
| Polyethylene low density | BTX aromatics, mono- & dimethylbenzene, trimethylbenzene, indane, indene, methylidenes, naphthalene, alkylated naphthalene, acenaphthylene, acenaphthene, fluorene, C4–C60 hydrocarbon: 1-alkenes (>C4) and others | Williams and Williams, 1999; Wampler, 1989 |
| Polyethylene high density | BTX aromatics, cyclohexene, methyl cyclopentene, C4–C60 hydrocarbon: 1-alkenes (>C4) and others (mainly oligomers, with additional amounts of C10+4x oligomers) | Wampler, 1989; Jung et al., 2010b |
| Polypropylene | BTX aromatics, 2-methyl-1-pentene, 3-methylcyclopentene, higher yield of C4–C13 hydrocarbons (mainly branched), lower yield of >C13 hydrocarbon (mainly branched), Ethylbenzenes, Indene, Biphenyl | Wampler, 1989; Jung et al., 2010b |
| Polystyrene | Styrene, ethylbenzene, cumene, propylbenzene, 2-ethyltoluene, naphthalene, diphenylmethane, anthracene, 1,2-diphenylethane, 2,2-diphenylpropane, 1,3-diphenylpropane, phenylnaphthalene, diphenylbenzene, triphenylbenzene, BTX aromatics | Onwudili et al., 2009 |
| Polycarbonate | Phenol, 4-methyl-phenol, 2-methyl-phenol, 4-ethyl-phenol, 4-(1-methylethyl)-phenol, 4-isopropenyl-phenol, 4-(1-methyl-1-phenylethyl)-phenol, 4,4'-(1-ethylidene)bis-phenol, 4,4'-(1-methylethylidene)bis-phenol, aromatic and aliphatic hydrocarbons | Antonakou et al., 2014 |
| Polymethyl methacrylate | Methyl methacrylate (>92 wt%) | Panda et al., 2010; Punkkinen et al., 2017 |
| Polyamide 6 | Caprolactam (up to 100 wt%) | Lehrle et al., 2000; Punkkinen et al., 2017 |
Industrial pyrolysis processes for the thermal treatment of municipal waste have not been able to gain importance due to the significantly lower technology readiness level compared to waste incineration and the higher treatment costs caused by the more complex technical design (Gleis et al., 2018). New developments in legislation make pyrolysis an interesting pre-treatment of specific waste fractions to yield a condensate providing access to a carbon source suitable for further processing. In terms of circular economy, chemical recycling with pyrolysis as pre-treatment could prove to be a method of closing the gap for plastics that cannot be mechanically recycled due to their respective molecular structure, e.g., thermosets. Again, the viable end-of-life scenarios have to take into account the already implemented value chains, thereby evaluating the replacement potentials (Meys et al., 2020). Additionally, pyrolysis could be a possibility to use specific waste fractions as raw material and generate valuable chemicals and precursors for biotechnological upcycling and subsequent use in various industrial and commercial applications.
3. Monomers as substrates for microbes
The previously described methods for plastic depolymerization are effective ways for making plastics accessible for microbial digestion. In many microbes, we can exploit or engineer the bow-tie structure of metabolism (Fig. 1(Sudarsan et al., 2014; Becker and Wittmann, 2020; Jovanovic et al., 2021),) that enables the use of alternative substrates to fuel the central carbon metabolism, while finally producing, besides biomass, a single product of choice. In the following, we describe metabolic pathways and microbes capable of metabolizing the main monomers identified above. While some monomers (such as SA and octanoic acids) are catabolized in common central carbon metabolic pathways, others require highly specific routes and enzymes, which are not available ubiquitously. To reduce the high number of monomers emanating from the above enumerated depolymerization methods, we focus on the compounds specified in Table 4. For some monomers, we identified more generic proxies, as also stated in the table. In the following, the monomers are sorted by their assimilation into central carbon metabolism (upper glycolysis, β-oxidation, TCA cycle, aromatics degradation pathways).
Table 4. Monomers arising from plastic degradation and the respective polymers they are generated from. Some monomers are used as proxies for similar molecules in the subsequent section on microbial metabolization.
| Representative monomers | Polymers | Proxy for |
|---|---|---|
| Ethylene glycol | PET, PEF, PUR | – |
| D-lactic acid | PLA | – |
| Styrene | PE, PP, PS, PC | Aromatics |
| 3-Hydroxyoctanoic acid | PHA, PE, PP, PUR | 3-Hydroxyalkanoic acids, C4–C13 hydrocarbons |
| Hexadecan(oat)e | PE, PP, PC | Aliphatics, >C14 hydrocarbons |
| 2,4-Toluenediamine | PUR | 4,4′-Methylene dianiline |
| Caprolactam | PA | 6-Aminohexanoate, hexamethylenediamine |
| Adipic acid | PBAT, PUR, PA | Azelaic acid, Sebacic acid, Suberic acid |
| Methyl methacrylic acid | PMMA | – |
| 1,4-Butanediol | PBS, PBAT, PUR | – |
| Succinic acid | PBS | – |
| 2,5-Furandicarboxylic acid | PEF | – |
| Terephthalic acid | PBAT, PET | – |
| Phenol | PE, PP, PS, PC | Aromatics |
One specific microbe shown to feature high potential for valorization of many monomers from plastic depolymerization is P. putida. This Gram-negative gamma proteobacterium has a very versatile metabolism, enabling the consumption of many different carbon sources. Pseudomonads have so far been engineered to metabolize the PET monomers EG (Li et al., 2019) and TPA (Kenny et al., 2012), as well as some PUR monomers such as AA (Ackermann et al., 2021), 1,4-BDO (Li et al., 2020), and even 2,4-toluenediamine (TDA) (Espinosa et al., 2020). With the example of P. putida's metabolic network, catabolic pathways of the here described monomers are shown in Fig. 2.
 Fig. 2. Metabolization of monomers from plastic depolymerization. The metabolic network of P. putida is shown including central carbon metabolism (CCM, blue) as well as some secondary pathways (Entner Doudoroff pathway – brown, β-oxidation - green). The entry points of the metabolization routes of plastic monomers into the central carbon metabolism are shown in yellow. For some monomers, alternative routes exist (see supplementary information “SI Table” for details), but are not shown for a better overview.
Fig. 2. Metabolization of monomers from plastic depolymerization. The metabolic network of P. putida is shown including central carbon metabolism (CCM, blue) as well as some secondary pathways (Entner Doudoroff pathway – brown, β-oxidation - green). The entry points of the metabolization routes of plastic monomers into the central carbon metabolism are shown in yellow. For some monomers, alternative routes exist (see supplementary information “SI Table” for details), but are not shown for a better overview.3.1. Ethylene glycol (EG)
EG was first synthesized in 1856 by Charles-Adolphe Wurtz (1856). Nowadays, EG is produced from ethylene, which in turn originates from petrochemicalresources via steam cracking of ethane (Pang et al., 2016). A commercial bio-based route via ethanol has also been established (Harmsen et al., 2014). In 2019, about 42 Mt of EG were produced worldwide (Garside, 2020), with different applications in a wide range of polyesters (Yue et al., 2012).
So far, various organisms have been described that can use EG as the sole source of carbon and energy under both aerobic and anaerobic conditions (Fincher and Payne, 1962; Gaston and Stadtman, 1963). As has been demonstrated for a variety of different microbes, metabolization happens through the sequential oxidation of EG to glyoxylate (KEGG, R00476) via the intermediates glycolaldehyde (KEGG, R01781) and glycolate (KEGG, R01333) (Mückschel et al., 2012). In recent years, efficient metabolization of EG has been implemented in P. putida, either by metabolic engineering or by laboratory evolution (Li et al., 2019; Franden et al., 2018). A bottleneck in the above-described sequential oxidation was observed to be the final step from glycolate to glyoxylate. Overexpression of the gene coding for the membrane anchored oxidase GlcDEF removed this restriction. Further, the oxidation reactions should be balanced such that the toxic aldehyde intermediates glycolaldehyde and glyoxal do not accumulate. Another bacterium that has recently been engineered to utilize EG as carbon source was Escherichia coli (Pandit et al., 2021; Panda et al., 2021).
For further metabolization of glyoxylate as sole carbon source, two main options exist: 1) Routes only generating redox equivalent and CO2 and 2) routes that allow the usage of glyoxylate as carbon source. The first being the utilization of glyoxylate by the AceA (KEGG, EC 4.1.3.1) or GlcB (KEGG, EC 2.3.3.9) enzymes involved in the glyoxylate shunt (Li et al., 2019). This route can, at least in part, enable utilization of glyoxylate as carbon source, but only if a co-substrate is provided. For the utilization as sole carbon source, it was discovered that P. putida KT2440 features the glyoxylate carboligase (Gcl) enzyme (KEGG, EC 4.1.1.47), converting two glyoxylate molecules into tartronate semialdehyde and CO2 (KEGG, R00013). The former is converted to glycerate, either directly (KEGG, R01745) or via hydroxypyruvate (KEGG, R01394, R01388), and subsequently to the glycolysis intermediate 2-phosphoglycerate (KEGG, R08572) (Franden et al., 2018).
The described pathway from glyoxylate to a central carbon metabolite has a yield of 0.75 mol/mol, as one CO2 molecule is released. An alternative pathway for EG assimilation without carbon loss has been described by Wiegant et al. (Wiegant and de Bont, 1980). Here, a diol dehydratase generates acetaldehyde from EG, which is converted to acetyl-CoA via acetate, but the feasibility of this reaction for metabolic engineering purposes is questionable. The dehydratase is oxygen sensitive, requires vitamin B12, and in its native host, it is part of a much larger context of accessory enzymes and a protein microcompartment that hinder its functional expression in other organisms (Crowley et al., 2010). In a more recent approach, Lu et al. (2019) used enzyme engineering to construct a synthetic pathway from C1 substrates to acetyl-CoA, which can also be utilized for 100% carbon-efficient EG metabolization. Glycolaldehyde is converted to acetyl-phosphate, which is transformed to acetyl-CoA. In a similar approach, Scheffen et al. (2021) enabled the conversion of glycolate and CO2 to glycerate.
3.2. Lactic acid (LA)
LA has been discovered in history three times in different organisms. It was first isolated by Carl Wilhelm Scheele in 1780 from milk (Parks et al., 2020) and in 1808 discovered in muscles after exertion (Kompanje et al., 2007). Finally, in 1813, Henri Braconnot found out that LA was present in fermented media (Benninga, 1990). Today, an estimated 1.5 Mt of LA are produced (Rodrigues et al., 2017). About 90% is derived from microbial fermentations, traditionally using different Lactobacillus strains (Hofvendahl and Hahn–Hägerdal, 2000), while nowadays pH-tolerant bacteria and yeast are used for the production of both stereoisomers, of which L-lactate is dominating the market.
Metabolization of LA involves a single step to form the central metabolite pyruvate (KEGG, R00704) via the lactate dehydrogenase (LDH) (KEGG, EC 1.1.1.28). LDH is an enzyme found in nearly all living cells (Tang et al., 2017). However, in general, LDH are utilized for the synthesis of LA. The reverse reaction (i.e., LA oxidation) is commonly considered to be only catalyzed by NAD-independent LDH (Garvie, 1980).