1. Introduction
Gold extractive metallurgy has arguably been the subject of more studies and publications than any other metal. Pyrometallurgical processing and physical separation methods were the earliest forms of gold processing, followed by mercury amalgamation in the more recent past, but these were overtaken by use of cyanide as chemical lixiviant. Early industrial mining applications using cyanide date back to 1887 (Maclaurin, 1895; Gray and McLachlen, 1933). Cyanide use has become ubiquitous, with about 80% of global gold extraction being facilitated by this lixiviant, despite the large number of other lixiviants that have been proposed (Aylmore, 2016). Globally, more than a billion tons of ore per year are treated using cyanide (Habashi, 2016). In addition to the beneficial gold-lixiviant features of cyanide, its associated hazards also became known in the 19th Century, and with it the critical importance of pH control. Lime is typically the most cost-efficient reagent to achieve this. The natural abundance of limestone from which lime is produced, the cost competitiveness of lime and its chemical features have entrenched it as a critical reagent, alongside cyanide, in the gold processing industry. A significant body of scientific literature has been devoted to the industrial use of cyanide in gold processing (Adams, 1990; Marsden and House, 2006), in contrast to the dearth of publications on the use of lime in this important industry. This review, therefore, explicitly focuses on lime use in gold processing applications. The large variety of gold processing methods necessitated limiting the scope of this review to current industrial applications and to cyanide as the dominant gold lixiviant.
2. Lime reagent formats and features
Lime is the generic term used to describe various formats of the reagent (National Lime Association, 1995; du Plessis et al., 2021). Formats include: quicklime – a dry reagent with calcium oxide (CaO) as the main component, produced from the calcination of limestone containing calcium carbonate (CaCO3); hydrate or hydrated lime – a dry reagent comprising mainly Ca(OH)2, produced from the stoichiometric hydration of quicklime with water; and slaked lime or milk of lime – a slurry of Ca(OH)2 particles typically in the range of 10–25 %wt produced from slaking quicklime in a stoichiometric excess of water, although specialty product formulations may contain up to 45 %wt solids content.
Quicklime (with active ingredient CaO), the starting reagent material for all other forms of lime reagent, is produced by calcination of limestone (CaCO3) at temperatures ranging from 900 to 1300 °C (Kumar et al., 2007) using a variety of kiln types and fuel sources (Piringer, 2017). Calcination is typically outsourced to lime producing companies, usually located some distance from the mining location. The calcination conditions, as well as the fuel used as energy source, significantly influence the properties of the resulting quicklime (Boynton, 1980). These factors include the impact on quicklime porosity, reactivity, slaking characteristics, and ultimately the Ca(OH)2 particle size generated after reacting with water (Zanin et al., 2019). The remote locations of most mine sites make transport logistics a significant cost component, particularly given the fairly low cost of the lime raw material at the point of production. Quicklime is typically used for long distance transport because of its lower mass per unit of reactive ingredient, compared to hydrate and slaked formats.
3. Quicklime composition
Several similar international standard methods have been developed and are widely used to characterize lime reagents (AS 4489.6.1-1997; ASTM C25 – 19; EN, 2017). The parameter ranges of typical industrial quicklimes are illustrated in Table 1, and the relevance of each briefly discussed.
Table 1. Parameter ranges of typical industrial quicklime.
| Parameter | Range (%wt) |
|---|---|
| Total CaO content | 80–95 |
| CaO available | 75–92 |
| MgO | 1–5 |
| SiO2 | 1–5 |
| Al2O3 | 0.07–0.7 |
| Fe2O3 | 0.08–0.4 |
| SO3 | 0.1–1.3 |
| Loss on ignition (LOI) | 0.5–2.5 |
| CO2 | 0.4–2.0 |
The total CaO content is typically determined by X-ray fluorescence spectroscopy (XRF) and is indicative of the calcium present in different mineral forms, mainly as quicklime (CaO). This metric is distinguished from available CaO, determined by acid titration of a slaked sample. The available CaO value is an indicator of the active reagent component that is available for the target reaction and is, therefore, less than the total CaO. A portion of the total CaO is unreactive, with this proportion increasing with calcination time and temperature (Boynton, 1980; Zanin et al., 2019). Some magnesium is unavoidably present in virtually all quicklime, because of the inherent association of magnesium with calcium in limestone. The magnesium content of limestone is of lesser reagent value in gold processing than the calcium content. This is for a number of reasons: (1) MgO in lime is significantly less reactive than CaO; (2) MgO cannot raise solution pH above 10.5, compared to 12.4 in the case of CaO; and (3) the reaction product of MgO in most gold processing applications is soluble MgSO4, which acts as a buffer against pH increases above 10.5, thereby increasing overall lime consumption. The silica and iron content of lime is typically low and inconsequential in gold processing, as it mainly remains inert. Most of the sulfur content originates from the calcination fuel used, rather than from the limestone. Sulfur containing fuels, therefore, increase the sulfur content of the lime. An elevated sulfur content of lime, particularly if it exceeds 0.5 %wt may have a detrimental effect during slaking of quicklime, as it has a coarsening effect on the slaked Ca(OH)2 particles (Potgieter et al., 2003; du Plessis et al., 2020). Loss on ignition is defined as the weight loss due to thermal decomposition from heating to a defined temperature. For lime it typically covers two different metrics: Most commonly, it describes the loss on ignition up to 950 °C, i.e. the weight loss is caused by the decomposition of both the calcium carbonate and calcium hydroxide in the sample to quicklime (CaO). The other metric is the loss on ignition up to 550 °C, caused by the decomposition of calcium hydroxide to CaO. This metric becomes important when a quicklime shipment has been exposed during transport, handling or storage and has reacted with air humidity. This reaction decreases quicklime reactivity, as well as the available CaO to a minor degree. A lime supplier will typically only provide loss on ignition values, upon heating to 950 °C, on the fresh quicklime shipment leaving its production site. From the loss on ignition value (to 950 °C), the amount of residual CaCO3 (typically unburned limestone) can be calculated (EN12485:2017; ASTM C25-17). As the loss on ignition (to 950 °C), or the difference between the loss-on-ignition up to 950 °C and 550 °C for “air-slaked” lime, is equivalent to the amount of CO2 liberated by decomposition of calcium carbonate, this value is commonly also referred to as “CO2 content”. In the same fashion, the loss of ignition upon heating to 550 °C is commonly referred to as the “H2O content”, i.e. the water vapor liberated by decomposition of calcium hydroxide. Analytical methods for characterizing lime, also include determination of quicklime reactivity (not shown in Table 1, because of different determination methods used in different regions), measured by a temperature increase upon reaction with water. A rapid increase in temperature indicates a higher reactivity. Reactivity is influenced by a range of factors, including the quicklime purity and the calcination temperature (Boynton, 1980; Oates, 2008; Zanin et al., 2019). Limestone calcination at 900⁰C results in highly reactive quicklime compared to ultra-low reactivity from calcination at 1,300 °C. Reactivity loss from calcination at increased temperatures is typically (Boynton, 1980) explained by sintering and grain growth and thus loss of reactive internal surface area. As grain growth is thought to be driven by surface diffusion during calcination, increased temperatures accelerate this process.
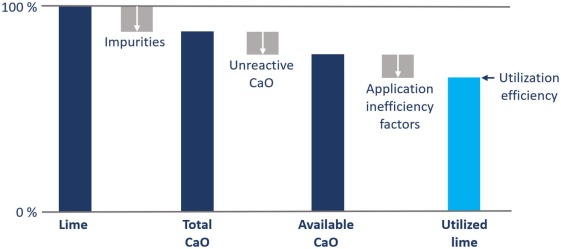
Lime reagent utilization efficiency can be defined as the mass percentage of the lime reagent participating in the target reaction(s). Utilization efficiency is not generally explicitly determined in gold processing operations, although it is an important consideration when comparing lime reagents and can readily be determined under laboratory conditions, using relevant proxy reactions (Ineich et al., 2017). It should be noted that although the available CaO content of lime provides an indication of potential active reagent, this metric is determined for quicklime and does not take into consideration the factors that influence slaking and thus the properties of the Ca(OH)2 particles. It is the properties of hydrated Ca(OH)2 particles, rather than the starting quicklime, that determine utilization efficiency. Lime utilization efficiency is, therefore, a combination of lime impurities, unreactive CaO, application inefficiencies and properties of the Ca(OH)2 particles (Fig. 1).
 Fig. 1
Fig. 14. Dry quicklime versus slaked lime reagent use
Lime reagent dosing methods in gold processing are typically dependent on the extent of acid consuming reactions and the accuracy of pH control required. The simplest dosing format is where dry quicklime (of particle size < 25 mm) is directly applied to ore on a conveyor-belt ball mill feed via a dosing screw attached to a lime silo. The dosing screw feed rate is controlled by a feedback mechanism based on the pH measurement in the cyanidation process unit. Such dosing methods are typically applied in scenarios where the lime consumption demand of processed ore is relatively low (i.e. < 2 kg per ton of ore treated), predictable and stable. Lime dosing via this method is based on the rationale that the ball mill provides sufficient agitation and residence time to achieve slaking (i.e. hydration) of the quicklime into Ca(OH)2. A similar dosing method is applied to gold heap leaching operations where dry quicklime is dosed directly from a silo, via a screw feeder, onto the ore on the back of a truck before the ore is stacked onto a heap (Kappes, 2002). The latter method does not provide for precise pH control or adjusting pH after lime has been added to the ore. Despite these limitations, such dry addition methods are relatively simple to implement with minimal operational or capital cost. In most gold processing operations and applications, however, dry quicklime is slaked resulting in a suspension of Ca(OH)2 particles that can be pumped and dosed into multiple process operations for flexible and accurate pH control. Slaking can be achieved in a variety of slaking reactors including ball mill slakers, detention slakers, and stirred tank slakers (Oates, 1998).
5. Ca(OH)2 as active reagent format
A feature of virtually all non-pyrometallurgical gold processing is that it occurs in aqueous media. In such conditions, Ca(OH)2 is always the active reagent component of lime, regardless of the format in which lime is dosed into the process. Even in scenarios where dry quicklime is used, the CaO does not directly react in target reactions. Instead, CaO first reacts with water (hydrates) to form slightly soluble (∼1.4 g.l−1) Ca(OH)2 particles, or more precisely Ca2+ and OH− (Oates, 2008). It is the soluble calcium (Ca2+) and hydroxide ions (OH−), rather than the solid Ca(OH)2, that participate in target reactions (Giles et al., 1993). Dissolution of Ca2+ and OH− from the surface of solid Ca(OH)2 particles is the limiting step in the reaction (Johannsen and Rademacher, 1999). Finer Ca(OH)2 particles result in improved kinetics and utilization efficiency, compared to larger Ca(OH)2 particles. Typically, a Ca(OH)2 particle size with d50 below 15 µm kinetically performs optimally, even though it is rarely explicitly measured in gold processing plants. Ca(OH)2 particle size is mainly determined by the following factors (Ineich et al., 2017):
-
•
Reactivity of the lime (influenced by the calcination process conditions and fuel), with more reactive lime typically resulting in finer Ca(OH)2 particles (National Lime Association, 1995).
-
•
Soluble elements contained in the slaking water, e.g. high concentrations (>500 mg.l−1) of sulfate in the water will coarsen the milk of lime (Potgieter et al., 2003).
-
•
Operating temperature of the slaker, i.e. higher slaking temperatures (65–85 °C) generally result in finer Ca(OH)2 particles (Zinck and Aube, 2000). This phenomenon is particularly relevant for low reactivity lime, but less relevant for higher reactivity lime. For a continuous production process, the slaking temperature is primarily controlled by the lime-to-water feed ratio of the slaker (typically at mass ratio of 1:8 to 1:4) and the reactivity of the lime.
-
•
Extent of mechanical abrasion or comminution during or after slaking. Some slakers, for example, make use of comminution methods (as in ball mill slakers) to reduce the particle size of the slaked lime. This is particularly used in scenarios, where other factors would otherwise result in coarse Ca(OH)2 (Gärtner and Diaz Chavez, 2019).
6. Cyanide as driver for lime use
The use of cyanide as the predominant gold lixiviant reagent, Reaction (1), is the key driver for the use of lime in gold processing (Perry et al., 1999). Cyanide, commonly used in the form of sodium cyanide (at solution concentrations ranging from 200 to 600 mg.l−1 or dosages of 0.1 to 1.0 kg per ton of ore (in the case of non-refractory ore), acts as a lixiviant for gold in the presence of oxygen, where oxygen facilitates oxidation of zero valance metallic gold (Au0) into monovalent gold (Au+), with which cyanide forms a stable complex [Au(CN)2]−, Reaction (1), referred to as aurocyanide. Calcium cyanide is less commonly used and then only where production occurs close to the point of use. This is because dry powdered cyanide is prone to hydrolysis when in contact with most air, which causes release of HCN gas and the use of slurries/solutions limit the economical transport distances of cyanided in this format.
The cyanide reagent used in gold processing significantly exceeds the stoichiometric gold content in the ore/concentrate being treated. This is because most of the added cyanide is either consumed by unwanted side-reactions (Bakatula and Tutu, 2016) and/or rendered ineffective due to the pH-dependent HCN/CN− speciation of cyanide illustrated in Fig. 2a, with a pKa of 9.2, Reaction (2). Importantly, CN− is the desirable species for gold cyanidation leaching, thus requiring the pH of the solution to be maintained, preferably above pH 10.5, where > 90% of the cyanide is present as CN−, to enable the gold dissolution reaction. Below this pH, soluble cyanide is increasingly present as hydrogen cyanide (HCN), which is ineffective as a leaching agent and prone to volatilization as a highly toxic and hazardous gas (Perry et al., 1999). Chloride in saline gold processing solutions, ubiquitous in the Western Australian gold fields (Turner et al., 1994; Ali and Turner, 2004), causes a shift in equilibrium between HCN and CN− towards lower pH values (Verhoeven et al., 1990) (Fig. 2b).
 Fig. 2
Fig. 2The cost of cyanide is typically a major expense for many gold operations. The use of lime, therefore, enhances the utilization efficiency of cyanide via pH modification. In scenarios where process solutions are buffered, often with dissolved magnesium (detailed in the next section), the lime reagent requirement to attain a pH level higher than 10.5 becomes a high operating cost component. Under such conditions, operational lime-cyanide trade-off decisions have to be made weighing up the cost of: (a) operating at a pH level of 10.5 with increased lime reagent to increase the cyanide utilization efficiency (and thereby the cost efficiency) compared to (b) operating at a pH of, say, 9 (below the buffering capacity of magnesium) saving on lime reagent but increasing cyanide consumption due to lower utilization efficiency.
7. The main alkali consumption reactions in gold processing circuits
In the absence of alkali consuming reactions, the relationship between Ca(OH)2 concentration and solution pH is illustrated in Fig. 3. Empirically the relationship can be estimated from reactions (3), (4), where Ca(OH)2 concentration is in grams per liter, within the pH range 7–12 and soluble Ca(OH)2 concentration < 1 g.l−1. These empirical equations generally fit the Ca(OH)2 versus pH titration curve better than the theoretical calculation based on the OH− concentration, in unbuffered solutions (Lhoist internal data).
 Fig. 3
Fig. 3In addition to raising the pH to the operational setpoint for optimal cyanidation, lime is also required to neutralize and counter acid-generating reactions or to overcome the effects of dissolved pH-buffering elements. The main lime consuming reactions that, in combination, contribute to the overall lime consumption of gold processing plants, are briefly discussed:
Magnesium pH buffering – Magnesium is an abundant element contained in gangue ore minerals (Chryssoulis and McMullen, 2016) and in process solutions. Magnesium is solubilized at pH levels below 9.5 and particularly under more acidic conditions. Conditions conducive to magnesium dissolution may occur in tailings storage facilities, from which the return water is used as process water, and within the process circuit before sufficient lime reagent has been added to achieve the target pH for cyanidation. Once magnesium is in solution, usually with sulfate as the counter-anion, it buffers the pH and consumes alkali when upward control of pH is required. This soluble magnesium starts reacting with lime, Reactions (5a) and (5b), from pH 9.3 to 9.5, and continues to consume lime until a pH of 10.5 is reached (Ineich et al., 2017). Stoichiometrically, each ton of dissolved Mg2+, will consume 3.1 tons of Ca(OH)2 (equivalent to 2.3 ton of CaO). An important consideration for plant operators is to conduct an inventory of dissolved magnesium input into the entire plant water circuit. If magnesium inputs into the water circuit can be minimized, the lime-cyanide trade-off consideration mentioned earlier may favour an operational pH > 10.5. Initial lime consumption in the process circuit may be high, as the initial process water inventory of magnesium is precipitated from solution, but would subsequently be reduced to the extent that further magnesium input into the water circuit can be minimized. If soluble magnesium is continuously and unavoidably entering the solution circuit at high concentrations, the resulting high lime consumption it would induce (as well as the increased viscosity due to precipitation of large quantities of Mg(OH)2 and CaSO4·2H2O) may force a plant to operate at pH values in the range 9.0–9.2 (Costello, 1986) to avoid this reaction, and incur higher cyanide consumption instead.
Acid generation from pyrite oxidation – The presence of pyrite (FeS2) or other sulfide minerals results in acid generation when exposed to water and oxygen, Reaction (6). This reaction may occur within the processing plant, in waste dumps or in tailings storage facilities (Dold, 2014), and may be significantly accelerated by the presence of bio-catalytic microbes (Duncan et al., 1967; Livesey-Goldblatt, 1986). The acid from this reaction is neutralized by lime resulting in the formation of gypsum precipitate, Reaction (7). Stoichiometrically, each ton of oxidized pyrite requires 1.24 tons of Ca(OH)2 (equivalent to 0.93 ton of CaO). The fact that gypsum precipitates in the process means that both the sulfate anion in sulfuric-acid and alkali cation are removed from solution, thus preventing their accumulation in the circuit. This is an important distinguishing feature of using lime as alkali reagent. By comparison, the use of any sodium-based neutralizing reagent leaves the sodium soluble and without a chemical precipitation sink within the processing circuit. Sodium can, therefore, only exit the circuit via uncontrolled seepage from tailings storage facilities (Zhang et al., 2020), unless deliberately removed from a side-stream by evaporation or reverse osmosis with the concentrate brines contained or treated, processes that are generally uneconomical for these applications. Apart from cost factors, the operational trade-off decision, between using calcium versus sodium alkali reagent (or other alkalis such as potassium or magnesium), is based on the following two options: (1) removal of most cations and anions from solution and dealing with the disposal of precipitated gypsum and metal hydroxides, compared with (2) dealing with soluble sodium sulfate as reaction product, that cannot readily be removed from solution. Pyrrhotite also occurs in some gold ores and is a significantly more reactive iron sulfide mineral than pyrite. The net acid generation, and therefore the lime consumption effect of its oxidation, is dependent upon a number of complex factors, including the ratio of contained iron to sulfur, the extent of oxidation of iron and sulfur, and the nature of precipitation products (Belzile et al., 2004).
Reactions with clays contained in laterites - Substantial gold mineralization occurs in some relatively shallow weathered tropical laterites (Colin and Vieillard, 1991; Larizzatti et al., 2008; Kioe-A-Sen et al., 2016). Due to extensive weathering over geological time, such laterites contain an abundance of hydrated alumina and silica, often in clay-like format (Obonyo et al., 2014). The reactions of hydrated lime with clays contained in weathered laterites is well known in civil engineering soil stabilization applications (Little, 1987; Transportation Research Board, 1987). In the context of gold processing, the available time frame determines the most relevant lime consuming reactions with this ore material. The relatively short time period (i.e. hours), from lime addition to cyanidation leaching occurring in a gold processing plant, causes cation exchange capacity interactions to be the most prominent lime consuming phenomenon (Little, 1987). When lime is added to such clay-containing laterite slurries, the divalent calcium cation displaces most other cations already adsorbed onto the negatively charged clay surfaces. Ca2+ displaces all of the cations to its left in the Lyotropic series (Little, 1987): Li+ < Na+ < H+ < K+ <NH4+ ≪ Mg2+ < Ca2+ ≪ Al3+. Once adsorbed onto the clay surface, the calcium cations act to adsorb negatively charged hydroxyl anions. The net effect is that clay minerals adsorb both Ca2+ and OH− from solution, thus increasing the consumption of Ca(OH)2 added to the process. The clays contained in laterite gold ores effectively act as a physico chemical adsorption buffer against lime-induced pH increase to a level optimal for cyanidation leaching. Such ores have been reported to consume lime to levels as high as 12 kg per ton (IAMGOLD, 2018). Over extended time periods, as prevails in tailings storage facilities, pozzolanic-type reactions of residual lime with silica and alumina clay minerals may occur to form calcium silicate hydrate (CSH), CaO.SiO2·H2O, and calcium aluminate hydrate (CAH), CaO.Al2O3·H2O, respectively (Attoh-Okine, 1995; Firoozi et al., 2017; Saing et al., 2017).
Carbonation – The fact that the cyanidation reaction requires oxygen, Reaction (1), necessitates the introduction of either pure oxygen gas, active air sparging, or passive aeration in the case of gold heap leaching operations. The contained CO2 in air dissolves into solution from the liquid/air contact and reacts with lime to form calcium carbonate, Reaction (8), although this is the smallest contributor to overall lime reagent consumption. The product of this reaction is more relevant than the reagent consumption effect, because of the operational implications of calcium carbonate precipitate formation and very low solubility (<50 mg.l−1) (Plummer and Busenberg, 1982).
Calcium carbonate causes scaling effects that manifest most problematically on the surface of activated carbon, which is used to adsorb the aurocyanide complex from solution (discussed in more detail in the next section). General carbonate scaling in plant circuits may be addressed using anti-scalants such as polyether polyamino methylene phosphonates, polyacrylate or polyacrylate-polymaleate (Alfano and Boffardi, 1994).
8. Lime interaction with activated carbon
Adsorbing cyanide-solubilized gold onto activated carbon was a critically important development step in gold processing (Nicol et al., 1984a, Nicol et al., 1984b, Nicol and Fleming, 1984c) and played a significant role in expanding gold production. Lime and the consequences of its use, also play an important role in the performance of this process step and are briefly summarized.
Adsorption onto activated carbon – Cyanidation results in gold solubilized as negatively charged aurocyanide [AuCN2]−. Activated carbon has no surface charge and does not adsorb aurocyanide as a negatively charged gold-cyanide ion complex. The predominance of dissolved Ca2+ from the use of lime, despite the addition of cyanide in the sodium form, results in the formation, Reaction (9), of a weakly-soluble calcium aurocyanide non-ionic covalent coordination complex, Ca[Au(CN)2]2, that adsorbs onto the surface of activated carbon (Davidson and Sole, 2007). The degree to which the aurocyanide complexes are adsorbed follows the series Ca > Mg > Li > Na > K, with calcium aurocyanide most strongly adsorbed (Davidson, 1974). The reasons for this phenomenon could be inferred from fundamental studies conducted on highly ordered pyrolytic graphite (HOPG) (Poinen et al., 1998) that found Ca2+ to have a higher charge-to-size ratio than Na+ and K+, and a higher coulombic attraction for aurocyanide anions, once intercalated amongst the graphene layers.
Scaling on activated carbon – The precipitation of calcium carbonate, referred to earlier, Reaction (8), also occurs on activated carbon, causing surface fouling and reducing adsorption capacity (Davidson et al., 1979; Cevallos Toledo et al., 2020). Calcium carbonate scaling occurs on the outer surface of activated carbon particles, thereby preventing both adsorption and elution of gold from the internal meso- and micro pores (Fig. 4) that generally have a surface area exceeding 400 m2.g−1 (McDougall, 1991). Most commercial activated carbon has a specific surface area of 800 to 1200 m2.g−1, which is predominantly contained within micropores <2 nm in diameter. To counter this effect, a portion of the activated carbon inventory is continuously subjected to an acid washing step, to reverse the CaCO3 scaling effect. Acid washing is typically conducted using either hydrochloric or nitric acid at 3% concentration for 20 to 60 min at ambient temperatures, although some plants use more aggressive temperature conditions (i.e. 60 °C). In industrial practice, acid washing is commonly conducted before gold elution, i.e. with the aurocyanide present on the activated carbon (Stange, 1999). Acid washing makes the elution more efficient as it allows aurocyanide, adsorbed in the internal pores of the activated carbon, to be eluted for subsequent electrowinning.
 Fig. 4
Fig. 4One way to reduce calcium carbonate formation is to reduce carbon dioxide input by sparging pure oxygen rather than air into the cyanidation process, although at significant cost. An alternative is to use lime in a CO2 scrubbing system, Reaction (8), (Samari et al., 2020) prior to sparging the scrubbed air into the cyanidation reactor.
9. Lime use in gold processing flow sheets
The diversity of gold ores has given rise to many process flow sheet configurations (La Brooy et al., 1994). Lime reagent consumption is determined by the ore characteristics rather than the small fraction of contained gold. From a lime consumption perspective, hydrometallurgical gold processing can broadly be classified using three ore features: (1) gold grade, (2) copper mineral content and (3) pyrite content, as illustrated in Table 2, with typical lime consumption ranges given as a function of ore feed (Lhoist data). The combination of these features influences the lime consumption relative to gold production output. Each of the main processing flow sheet methods is discussed in more detail, with specific reference to lime consumption.
Table 2. A simplified empirical representation of the three main ore characteristics (gold grade, copper content and pyrite content, at high H, medium M or low L relative concentrations) influencing the choice of gold processing method and impact on typical lime consumption.
| Processing flow sheet method | Gold grade | Copper content | Pyrite content | Lime consumption (kg.t−1 of ore) |
|---|---|---|---|---|
| Simple tank leaching | H | L | L | 0.8 to 1.5 |
| Tank leaching with nuisance copper | H | M | L | 0.8 to 1.5 |
| Copper-gold sequential flotation | H | H | L | 0.2 to 0.8 |
| Conventional heap leaching | L | L | L | 1 to 5 |
| Refractory treatment processes | H | L | H | 2.5 to 10 |
9.1. Simple tank leaching
The simplest (and commonly occurring) flow sheet configuration for a free-milling ore is illustrated in Fig. 5. In most plant circuits, gravity-recoverable gold is catered for in the flow sheet, as this fraction of the gold incurs the least operational effort (Bath et al., 1973; McGrath et al., 2018; Lowes et al., 2020). The gravity recoverable gold process is not lime-intensive and is, therefore, not discussed here. This flow sheet (Fig. 5) is typical for ores with reasonable gold grade (i.e. > 1 g Au per ton ore) and have sufficiently low concentrations of iron and copper sulfide minerals that do not interfere with the processing in terms of cyanide consumption or copper dissolution. Lime is added, usually in dry quicklime form, to crushed ore via a screw feeder from a lime silo onto the conveyor belt feed of a ball mill. Lime dose rates are typically relatively low, i.e. 0.5 – 2 kg per ton of dry ore feed. Process water is also fed into the mill together with the crushed ore to facilitate milling at the appropriate solids concentration and viscosity that are suitable for direct cyanide leaching. This method of lime addition relies on lime slaking (i.e. hydration) occurring within the ball mill, where the residence time and attritioning effect is adequate for hydration of CaO to Ca(OH)2. In circuits that use dilute comminution, where the mill classification product solids content is too dilute for direct cyanidation, a thickening stage (not shown in Fig. 5) is used to increase the concentration to around 50 %wt (depending on viscosity limitations) before cyanidation. Some lime may be used to facilitate flocculation in the thickener (Stange, 1999), although such lime addition requires slaking for effective use. In the leaching circuit, the ore slurry, commonly referred to as “pulp”, is then subjected to leaching in a series of sequential agitated cyanidation tanks, to which cyanide is added and air is sparged into, to enable gold leaching (Adams, 1989). Once solubilized, gold adsorption occurs in a separate series of agitated tanks, in the case of carbon-in-pulp (CIP) processing. In these tanks, activated carbon flows counter-current to the flow direction of the pulp, with screens used to separate the mineral pulp from the gold-loaded activated carbon. In some scenarios the processing flow sheet may be configured to facilitate aurocyanide adsorption onto activated carbon simultaneous with (i.e. in the same vessels as) the leaching process, a process known as carbon-in-leach (CIL). This technique is used where particular characteristics of the ore increase the risk of gold loss by so-called preg-robbing or preg-borrowing (Dunne et al., 2012; Snyders et al., 2017). Preg-robbing refers to the phenomenon where cyanide-solubilized gold is adsorbed onto carbonaceous components contained in the mineral being treated, thereby “robbing” the pregnant solution of its soluble gold and rendering it unrecoverable. Some make-up lime may also be added to the cyanidation tanks for minor pH adjustment to maintain the target pH during cyanidation. Some plants perform milling in circumneutral pH (or the prevailing pH according to the water source) and add slaked lime slurry only to the cyanidation section.
 Fig. 5
Fig. 59.2. Nuisance copper treatment
All sulfide minerals react with cyanide, although to varying extents (Aghamirian, 1997; Deschênes et al., 2002), with copper minerals generally having a more detrimental impact on gold recovery. Some gold ores contain significant concentrations of copper minerals, (such as chalcocite, covellite, bornite, cuprite, chrysocolla, enargite, tetrahedrite, malachite and azurite) that are cyanide-soluble and significantly complicate the gold recovery (Kyle and Hefter, 2015), referred to as “nuisance copper”. The main reason for this “nuisance” is because such copper minerals often occur in much greater quantities (even if not in economic quantities) than gold and therefore result in significant cyanide consumption. The front-end flow sheet configuration for gold ores with nuisance copper is typically the same as illustrated in Fig. 5. Gravity gold is also often extracted, although the presence of native copper may also cause complications. Gold ores with nuisance copper differ from free-milling oxide ores in the back-end treatment of the cyanide-leached slurry. Cyanide reacts with copper minerals to form a range of copper–cyanide complexes, thereby consuming more cyanide than is necessary for gold leaching only. The copper–cyanide complexes are also quite stable under typical tailings conditions, when compared to free cyanide. These complexes compete with and reduce gold adsorption onto activated carbon (Dai and Breuer, 2009). This phenomenon also causes a range of operational problems including the increase of cyanide content in the leached tails and contamination of the final dore gold metal, amongst others (Shantz and Reich, 1978; Nguyen et al., 1997; Kyle and Hefter, 2015, Bakatula and Tutu, 2016). The sulfidization acidification recycle thickening (SART) process, is a way to deal with such copper and high cyanide consumption in gold processing circuits. SART has been incorporated in a variety of flow sheets, since its first installation at the Telfer gold mine in Western Australia, including tank and heap leaching operations (Barter et al., 2001; Adams et al., 2008; Stewart and Kappes, 2012; Estay et al., 2018). A simplified diagram of the main flow sheet components of SART processing of the cyanidation tailings solution is illustrated in Fig. 6, although several variants have been deployed. The most stable, and therefore most prevalent, copper–cyanide species at typical gold-process operational pH (i.e. 9 – 11) is [Cu(CN)3]2−, with copper in the cuprous valance state (Cu+). The first step of the SART process (i.e., sulfidization) is based on the high affinity of sulfide for copper, resulting in a very low solubility of copper sulfide precipitate (Ksp = 2.3 × 10−48) (Lewis, 2010; Estay, 2018). Using this phenomenon under acidic conditions (pH 4–5) allows for cyanide to be displaced from [Cu(CN)3]2− by sulfide reagents (NaSH or H2S) while cyanide is protonated to HCN, Reactions (10), (11), (Buisman and Dijkman, 2005; Adams et al., 2008). Acidification also promotes the breakage of the Cu-cyanide complex, as well as other weak acid dissociable (WAD) cyanide complexes with Zn, Ni, Ag, and Hg that may occur (Estay, 2018). The process results in the precipitation of Cu2S that is subjected to a thickening step to concentrate it, and releases cyanide back into solution as aqueous hydrogen cyanide (HCN). The thickened underflow, with the addition of NaOH, is filtered to produce the final Cu2S product. NaOH is used to increase the pH well above the 7.2 pKa of H2S/HS−, preferably to pH 10, to reduce the risk of H2S evolution from the Cu2S product. Lime can also be used for this purpose, but will result in gypsum co-precipitation in the product, that negatively affects the saleability of the Cu2S product.
 Fig. 6
Fig. 6The acid solution, from both the Cu2S thickener overflow and Cu2S filtration step, containing HCN is neutralized using lime, typically in an agitated reactor under slight negative pressure. This process step is used to both neutralize the acid, Reaction (7), and to increase the pH to fully convert HCN back to CN−, Reaction (12). The resulting gypsum (CaSO4·2H2O) is thickened, filtered and disposed to tailings, while the high pH treated solution, containing CN−, is recycled back to cyanidation. The main factors governing lime consumption are: (1) the quantity of acid used and (2) the buffering elements contained in solution. It should also be noted that the SART process contains two important gaseous hazards, HCN and H2S, both of which are pH species dependent, that require scrubbing (not shown in the diagram) and careful management.
9.3. Cyanide recovery by acidification, volatilization and recovery (AVR)
The AVR process, or different versions thereof, such as CRP (cyanide recovery process) and Cyanisorb, may be used to recover cyanide from process solutions with total cyanide concentrations exceeding 150 mg l−1, for re-use (Riveros et al., 1993; Stevenson et al., 1998). The process is generally more suitable to solutions rather than mineral slurries (Fleming, 2016). It can be used to recover cyanide from solution streams exiting the SART process, or as a standalone unit in scenarios where residual cyanide (i.e. after cyanidation leaching) is not significantly complexed by copper. In the AVR process, sulfuric acid is added to the alkaline cyanide containing solution to reduce the pH below 7, thereby converting contained cyanide to HCN. If cyanide is mainly present as WAD cyanide, a pH of 4 is required. The HCN is stripped or vapourised from the acid solution. The stripped acid solution is treated with lime reagent, to increase the pH to a suitable level for re-use within the processing plant and to precipitate soluble metals as hydroxides. The extent of lime use is dependent upon the acid addition used, as well as the quantity and extent of metal precipitation required. The HCN vapour is recovered by scrubbing into a NaOH solution. The cyanide concentration in the scrubbing solution can be built up to almost 100 g.l−1 as a maximum operational limit (Fleming, 2016). Lime may also be used in the HCN adsorption scrubbing unit, if sufficient measures are taken to overcome scaling. The net effect of using the AVR is that both WAD and free cyanides are separated and recovered from process solution streams for re-use in the cyanidation leaching step.
9.4. Sequential flotation recovery of copper and gold containing ore
Orebodies that contain both copper and gold in economic quantities, require flow sheet configurations tailored to their specific circumstances. The factors affecting the optimal configuration are: (1) the extent to which gold is associated with copper minerals or pyrite mineral, (2) the payable value of gold contained in the copper concentrate, and (3) country regulatory policies. The latter is relevant because regulation in some countries may compel companies to recover the full value of contained metals before exporting. The payable gold contained in copper concentrate is a function of the treatment charges – refining charges (TC-RC) that the mine operator can contractually secure for its copper concentrate (Malewski, 2016; Díaz-Borrego et al., 2019). Gold contained in copper concentrate is not payable at full value and, in some instances, attracts zero payable value. This provides a powerful incentive for mining companies to consider implementing process flow sheets that separately generate a copper concentrate as well as a gold value stream.
Copper-gold process flow sheets typically involve the combination of complex flotation circuits, combined with subsequent cyanidation leaching for gold recovery (Benson et al., 2007; Zheng et al., 2010; Medina and Anderson, 2020). In scenarios where gold is preferentially associated with pyrite, methods are required to separate copper minerals from gold-bearing pyrite. Though many different flow sheet combinations have been used commercially, a simplified block flow diagram of a typical flow sheet is illustrated in Fig. 7, showing the main lime addition points. Lime is added to the copper rougher flotation, typically to control the pH at approximately 9–10. Pyrite floats readily in acid and neutral conditions with xanthate as collector, while high pH values depress pyrite (Mu et al., 2016). Additionally, the high pH is also necessary because the oxidation of pyrite would otherwise cause a pH decrease in the copper rougher circuit, Reaction (6). Such a reduction in pH would facilitate some copper dissolution, causing copper activation of pyrite making it difficult to depress the pyrite (Zanin et al., 2019). In the Cu cleaner stage, the objective is to float and produce a high-grade copper concentrate, while the gold-containing pyrite is depressed (i.e. rejected to tails) (Bulatovic, 1997). This separation is pH dependent and typically requires a pH of 11, achieved by further lime addition (Akop, 2014; Zanin et al., 2019). The tails from this process report to the pyrite flotation circuit where pyrite is reactivated (i.e. the depression effect is reversed so that it will float), typically using potassium amyl xanthate (PAX). The gold-containing pyrite concentrate then reports to the cyanidation circuit for gold leaching and recovery (Benson et al., 2007; Akop, 2014).