ntroduction
It is estimated that ∼1/20 people suffer from one or more autoimmune diseases [1]. Furthermore, the ∼4 to 8% per annum increase in incidence of these diseases is a major concern (Table 1) [2]. Most autoimmune diseases are poorly managed by non-specific immunosuppressive drugs and there is currently no cure for the pathological mechanisms underlying these conditions.
Table 1. Increasing incidence and prevalence of immune mediated inflammatory diseases (IMIDs). The net %/year increase in categories of IMIDs including multiple sclerosis (MS), autoimmune hepatitis (AIH), inflammatory bowel disease (IBD), insulin dependent diabetes mellitus (IDDM), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). From Lerner et al. (2015). Prevalence of the diseases shown is from Hayter and Cook (2012)
| Organ/system | Significance | Mean net increase (%/year) | Countries of combined studies | Examples of IMIDs | Disease prevalence/105 |
|---|---|---|---|---|---|
| Neurological | <0.0001 | 3.7 ± 2.5 | Finland, Denmark, Norway, Italy, Spain | MS | 58 |
| Myasthenia gravis | 5.1 | ||||
| Narcolepsy | 30.6 | ||||
| Gastrointestinal | <0.0001 | 6.2 ± 11.5 | Denmark, Canada, Sweden, USA, Finland, Israel, Netherlands, UK, Czech, Scotland, Spain, Estonia, New Zealand | AIH type 1 | 16.9 |
| AIH type 2 | 3 | ||||
| IBD | 55 | ||||
| Celiac disease | 750 | ||||
| Pernicious anaemia | 151 | ||||
| Endocrine | 0.02 | 6.3 ± 4.2 | Brazil, Canada, Israel, Serbia, Europe, Canada, UK | Addison's disease | 14 |
| Rheumatic | 0.02 | 7.14 ± 1.5 | Graves’ disease | 629 | |
| Hashimoto's thyroiditis | 792 | ||||
| IDDM | 480 | ||||
| RA | 860 | ||||
| SLE | 32 |
Antigen-specific immunotherapy has been used for the control of allergy for over 100 years [3]. The mechanism by which allergic desensitisation controls allergy is not clear although it is known that T cells alter their cytokine secretion [4] and can support the generation of ‘blocking’ antibodies [5].
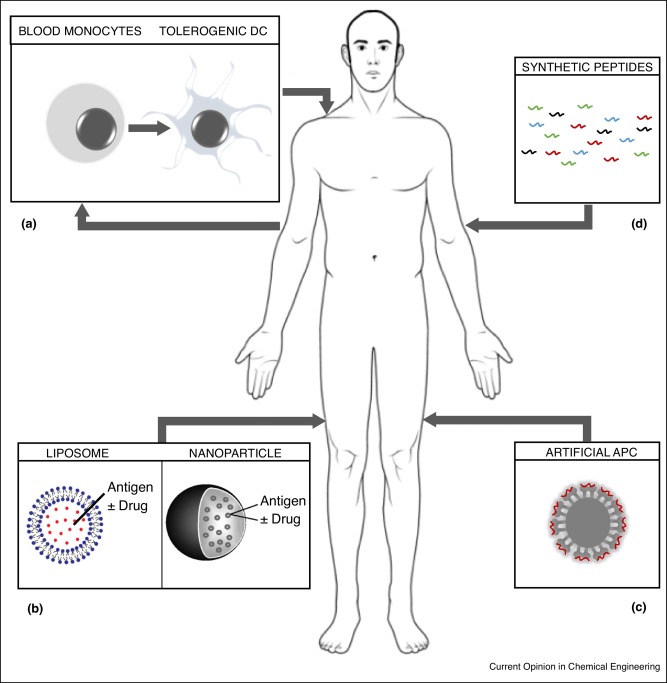
Antigen-specific immunotherapy is seen as the ‘holy grail’ for effective treatment of autoimmune diseases [6], it satisfies the need to induce protective immunity targeted to pathogenic T cells while avoiding non-specific immune suppression. The aim is to reinstate tolerance towards self-antigens while leaving the rest of the immune system capable of controlling infections and cancers. This review will describe recent advances in engineering self-antigens and creating novel platforms for their delivery to reinstate tolerance (Figure 1).
 Figure 1. Novel approaches to antigen-specific immunotherapy of autoimmune diseases: (a) Tolerogenic DCs are generated from the patient's blood-derived monocytes using various immunosuppressive agents to prevent DC maturation. Tolerogenic DC are pulsed with antigen and injected back into the patent. (b)Self-antigens are encapsulated or attached to liposomes or PGLA-nanoparticles. Drugs such as NFκB inhibitors may be added so as to prevent DC maturation in vivo. (c) Nanoparticles may be loaded with complexes derived from patients MHC II protein with self-antigen peptide pre-loaded. (d) Self-antigen derived T cell epitopes are optimised for solubility and MHC binding and injected directly without carrier or adjuvant. Details of these approaches and the outcome of their use are given in the text.
Figure 1. Novel approaches to antigen-specific immunotherapy of autoimmune diseases: (a) Tolerogenic DCs are generated from the patient's blood-derived monocytes using various immunosuppressive agents to prevent DC maturation. Tolerogenic DC are pulsed with antigen and injected back into the patent. (b)Self-antigens are encapsulated or attached to liposomes or PGLA-nanoparticles. Drugs such as NFκB inhibitors may be added so as to prevent DC maturation in vivo. (c) Nanoparticles may be loaded with complexes derived from patients MHC II protein with self-antigen peptide pre-loaded. (d) Self-antigen derived T cell epitopes are optimised for solubility and MHC binding and injected directly without carrier or adjuvant. Details of these approaches and the outcome of their use are given in the text.Tolerogenic DC for autoimmune diseases
The immune system generally remains dormant when faced with the myriad of self-antigens exposed throughout the body while responding rapidly and strongly to foreign antigens contained within infectious agents or the neoantigens resulting from mutations associated with cancer. In the case of infectious agents, the immune system is activated by recognition of pathogen-associated molecular patterns (PAMPS) from infectious agents. Dendritic cells (DCs) are the key antigen presenting cells (APCs) for activation of both CD4 and CD8 T-cells and are especially adapted to respond to PAMPS. Most importantly, Steinman and colleagues provided evidence that these cells are tolerogenic (see Box 1) in the immature/steady state and only promote strong immune responses when activated through ligation of receptors for PAMPS and other ‘danger’ signals [7]. Immature/steady state DC have low levels of the MHC class II and the costimulatory molecules (CD40, CD80 and CD86) required for effective activation of T cells. In addition, these immature DC secrete low levels of cytokines such as IL-12 that drive differentiation of effector T cells while having higher levels of anti-inflammatory cytokines such as IL-10. Antigens presented by tolerogenic DC are seen by T cells but this encounter results in T cell death, anergy or the adoption of a regulatory (Treg) phenotype.
Preferred features of tolerogenic dendritic cells
| A | Ability to process antigens or bind peptides directly via MHC class II at the cell surface |
|---|---|
| B | Reduced levels of class II MHC. Low levels of costimulatory molecules such as CD40, CD80 and CD86. Higher levels of inhibitory receptor ligands including PD-L1 |
| C | High ratio of anti-inflammatory (IL-10) versus pro-inflammatory (IL-12) cytokines |
| D | Ability to induce apoptotic cell death, anergy or the generation of regulatory T cells as a result of antigen presentation |
| E | Migration to lymphoid organs and stability in vivo |
| F | Resistance to DC maturation through ligation of toll-like receptors, CD40, etc. |
Various groups have sought means of preventing maturation of DC by treating immature cells with drugs, including vitamin D3, steroids and rapamycin [8] or by genetic manipulation. For example, DC can be treated with anti-sense oligonucleotides specific for CD40, CD80 and CD86 to create a tolerogenic population [9]. In a phase 1 study, 10 patients with type 1 diabetes were treated with DC either unmanipulated or engineered towards an immunosuppressive state ex vivo. The treatment was well tolerated although there was no significant change in relevant biomarkers.
Two studies of the use of tolerogenic DC for treatment of rheumatoid arthritis (RA) were published recently. Hilkens and Isaacs developed a method for engineering tolerogenic DC using dexamethasone, vitamin D3 and monophosphoryl lipid A [10]. The resulting cells display a tolerogenic phenotype, suppress T cell proliferation and secretion of interferon gamma and IL-17. Most importantly, DC treated in this way remained refractory to further challenge with pro-inflammatory cytokines in vitro. Equivalent cells pulsed with collagen type II had been shown to inhibit collagen-induced arthritis in mice [11]. On the basis of the success of this approach in mice, tolerogenic DCs were pulsed with autologous synovial fluid as a source of autoantigens and introduced into the inflamed joints of RA patients [12••]. The treatment was well tolerated with evidence of disease stabilisation in two individuals; however, no clinical or immunomodulatory effects were noted.
Thomas and colleagues have developed a similar approach using suppression of the NF-κB pathway as a means of generating tolerogenic DC. DCs modified with the NF-κB inhibitor, Bay11-7082, suppressed arthritis in an antigen-specific manner in mice [13]. Consequently, Benham and colleagues tested administration of Bay11-7082-treated DC pulsed with peptide antigens from citrullinated protein antigens [14••]. These antigens are found within inflamed joints of RA patients and anti-citrullinated protein antibodies (ACPA) are generated in the majority of RA patients [15]. DCs were administered once intradermally to 18 ACPA +ve patients. The treatment was well tolerated and there was evidence of an increase in the ratio of Treg/effector cells.
The studies described above demonstrate that it is possible to alter autoimmune phenomena in vivo by targeting antigens to tolerogenic DCs. This supports previous results from experimental models of multiple sclerosis (EAE), rheumatoid arthritis and type 1 diabetes [16]. Interventions in human disease are promising, well tolerated and a valid strategy for therapeutic tolerance in autoimmune disease.
Cell-free therapeutic approaches based on liposomes and red blood cells
These approaches have arisen from the original work of Miller and colleagues who showed the tolerogenic effect of splenocytes pulsed with antigen and then fixed with ethylene carbodiimide (ECDI) [17]. It was believed that these cells presented antigen but with disrupted costimulatory capacity following fixation. Subsequent studies showed, however, that the fixed APC could be replaced by antigen coupled directly to beads [18]. This revealed that cells pulsed with antigen and fixed with ECDI were carrying antigen to tolerogenic APC. Collective evidence implied that delivery of autoantigen to tolerogenic DC or the coincidental delivery of autoantigens with drugs designed to sustain the immature phenotype of DC in vivo could substitute for the transfer of tolerogenic DC (see Table 2). Pujol-Autonell created liposomes containing phosphatidylserine so as to mimic the surface phenotype of apoptotic cells, known to promote tolerogenic DC [7], and loaded them with antigenic peptide [19]. These liposomes reduced clinical symptoms of the mouse model of MS (EAE) when loaded with a myelin-antigen peptide and administered before disease induction. Kontos and colleagues followed a similar logic based on the clearance of senescent red blood cells [20]. They engineered antigen constructs to target antigens to erythrocyte cell surfaces after i.v. injection. This induced cell proliferation and apoptotic T-cell death in a cell transfer model of type 1 diabetes. It seems likely, however, that bystander suppressive mechanisms will be needed for effective immunotherapy of the many autoimmune diseases that are not limited to single antigens; this is not provided by such an apoptotic mechanism. With this in mind, Capini and colleagues created phosphatidycholine liposomes loaded with antigen and a lipophilic NF-κB inhibitor [21]. These liposomes are taken up by macrophages and DC in which they suppress NF-κB signalling. Treatment of mice with these liposomes led to an increase in the ratio of Treg:effector cells and produced a significant reduction of clinical disease in a mouse model of arthritis. These approaches show promise as alternatives to the generation and delivery of antigen-pulsed tolerogenic DCs.
Table 2. Carriers for antigen-specific immunotherapy of autoimmune diseases. This table gives examples of therapeutic approaches currently under development for treatment of autoimmune diseases. At this time, efficacy of these approaches has been demonstrated in mouse models of disease. There are no published reports of their use in human autoimmune diseases
| Carrier | Antigen-delivery mechanism | Immunosuppressive mechanism | Disease model tested |
|---|---|---|---|
| PS liposomes | Phosphatidylserine liposomes containing antigen | Uptake by and maintenance of a tolerogenic phenotype in DC. Increase in LAG-3 expression by T cells | Experimental autoimmune encephalomyelitis (EAE) in mouse |
| Red blood cell (RBC) | Antigen modified with RBC binding peptide or antibody fragment | T cell proliferation resulting in apoptosis of antigen-specific T cells | T cell transfer model of type 1 diabetes in mouse |
| PC liposomes and NFκB inhibitor | Phosphatidylcholine liposomes containing antigen and NFκB inhibitor | Uptake by macrophages and DC. Increase in Foxp3+ Treg cells and IL-10 secretion | Methylated bovine serum albumin induced arthritis in mouse |
| NP and RAPA | PLGA nanoparticles with antigen and rapamycin | Uptake by macrophages and DC. Increase in Foxp3+ Treg cells | EAE in mouse |
| NP | PLGA nanoparticles with antigen | Negatively charged particles are taken up by MARCO +ve monocytes and macrophages resulting in T cell anergy and increase in Treg cells | EAE in mouse |
| Artificial APC | MHC-peptide complexes were linked to dextran-coated or pegylated iron oxide nanoparticles | Direct recognition by antigen-specific T cells resulting in generation of Tr1-like regulatory T cells | EAE, collagen induced arthritis and type I diabetes in the mouse |
Cell-free therapeutic approaches base on nanoparticles
In an analogous approach to the work described by Capini et al., Kishimoto and colleagues developed an approach based on nanoparticles loaded with rapamycin. Rapamycin prevents maturation of DC and thereby holds the APC in a tolerogenic state [22]. Maldonado and colleagues originally combined protein or peptide antigens with rapamycin in biodegradable nanoparticles [23•]. This led to suppression of the autoimmune response to self-antigens in mouse models. More recently, Kishimoto and colleagues have shown that the approach depends on rapamycin being encapsulated within poly(lactic-co-glycolic acid) (PLGA) particles but their evidence suggests that the nanoparticles do not have to contain antigen [24]. This implies that the key feature of this approach is delivery of rapamycin to the DC and that co-administered antigen is taken up by the tolerogenic DC. However, this interpretation is challenged by the results of others showing that PLGA nanoparticles can be effective without inclusion of rapamycin or any other immunosuppressive agent [25•]. PLGA nanoparticles containing peptide antigens from myelin alone were shown to prevent and treat EAE in mice by upregulating PD-L1 on APCs and suppressing secretion of inflammatory cytokines by antigen-specific T cells. Antigen-specific immunotherapy with these nanoparticles depends on optimal engineering based on composition, size, charge and route of administration [26].
An alternative nanoparticle-based approach has been developed by Herkel and colleagues. This is based on the observation that expression of myelin basic protein (MBP) in liver tissue results in TGF-β dependent generation of Foxp3+Treg cells that suppress induction of EAE in a mouse model [27, 28]. Nanoparticles were designed to target liver sinusoidal endothelial cells (LSECs) since presentation of antigen by LSECs induces tolerance both CD4+ and CD8+cells [29, 30]. Nanoparticles were prepared from superparamagnetic iron oxidenanocrystals encapsulated in an amphiphilic polymer (poly(maleic anhydride-alt-1-octadecene)) and were shown to selectively target LSECs [31••]. Nanoparticles coated with peptides from MBP induced Foxp3+ Treg cells and reversed ongoing disease.
It is clear that nanoparticles can deliver antigen to endogenous tolerogenic pathways in the immune system thereby providing disease control without inclusion of immunosuppressive drugs. The major challenge is to identify which of these approaches induces bystander suppression since this will be essential for control of most autoimmune diseases.
Design of artificial APCs
A further development of the nanoparticle approach was described by Santamaria and colleagues [32••]. This creates artificial APC by coating nanoparticles with complexes of MHC class II molecules and antigenic peptides. These artificial APCs have optimal dimensions and spacing of MHC molecules for T cell receptor ligation but lack costimulatory molecules [33]. The artificial APCs directly modify antigen-specific Th1 cells by inducing Tr1-like cells capable of bystander suppression. Tr1 cells specific for one antigen mediate IL-10-dependent bystander suppression of T cell responses to other antigens when they are presented by the same APC [32••, 34]. The lack of costimulatory molecules on these artificial APC means that they interact with effector cells, such as Th1 cells, but do not modify the function of naïve T cells. As such, unlike other forms of antigen-specific immunotherapy that target tolerogenic DC, MHC-Ag bearing nanoparticles will suppress ongoing autoimmune diseases [32••] but will not serve as preventive treatments since they do not engage naïve T cells for which at least low level costimulatory signalling is required.
Design of synthetic peptides for antigen-specific immunotherapy
The ideal antigen-specific immunotherapy should allow repeated administration and induce a bystander suppressive mechanism in order to control the immune response to the range of antigens associated with autoimmune diseases. Is it necessary to use sophisticated delivery systems to produce this outcome? Weiner and colleagues originally provided evidence that autoimmune diseases could be controlled by oral delivery of autoantigens through induction of bystander suppression [35]. Whitacre and colleagues subsequently demonstrated, however, that high, repeated doses of protein were required for effective oral tolerance in the mouse model of multiple sclerosis. Such levels of protein would be impractical for use of this approach in humans [36] and would explain why trials of oral tolerance in human autoimmune diseases did not provide clinical benefit [37].
Further development of autoantigen-directed tolerance demonstrated that whole autoantigens could be replaced by synthetic peptides representing the T cell epitopes involved in disease, and that parenteral injection was more effective than mucosal administration of antigen [38••]. We have known for many years that administration of soluble peptide antigens both prevents and treats autoimmune conditions without the need for complex carriers [39, 40, 41]. Furthermore, we have defined key rules governing the design of effective therapeutic peptides (see Box 2).
Preferred features of tolerogenic synthetic peptides
| A | Peptides are highly soluble to avoid entrapment and destruction at site of injection |
|---|---|
| B | Peptides target tolerogenic DC |
| C | Peptides mimic naturally processed antigens when bound to MHC II |
| D | Peptides bind directly to MHC on tolerogenic DC |
| E | Repeated administration suppresses secretion of inflammatory cytokines by antigen-specific T cells while concomitantly inducing Treg cells and anti-inflammatory cytokines (IL-10) |
| F | Regulatory mechanism induced by peptide mediates bystander suppression |
| G | Administration of peptides derived from self-antigens is safe and well tolerated allowing repeated administration to patients |
Soluble peptide injection induces a state of anergy among antigen-specific T cells [42, 43], this coincides with suppression of pro-inflammatory cytokine production and upregulation of the anti-inflammatory cytokine IL-10 [41, 42]. The resulting Tr1-like cells mediate negative feedback regulation of the inflammatory immune response [44] and are capable of bystander suppression [34, 45].
The induction of Tr1-like cells with peptides correlates with changes in gene transcription in CD4+ T cells. Transcription factors, Maf, AhR and NFIL-3, known to support IL-10 transcription in Tr1 cells [46], are upregulated in T cells following peptide therapy [38••, 47]. Tr1-like cells isolated from treated mice upregulate inhibitory receptors such as PD-1, CTLA-4, LAG-3, TIM-3 and TIGIT [38••] while human Tr1 cells show variable levels of expression of LAG-3, CD49b [48] and TIM-3 [49].
Peptides must be optimised for solubility and MHC binding for effective antigen-specific immunotherapy. Studies with peptides from band 3 protein have shown that administration of insoluble peptide exacerbates haemolytic anaemia in the NZB mouse whereas soluble peptide prevents disease [50]. Solubility of peptides is improved by replacing hydrophobic with hydrophilic amino acids or adding charged amino acids to the N-termini or C-termini. Most importantly, peptides must be designed to bind MHC class II molecules in the correct conformation to induce effective tolerance. The processing of protein antigens by APC places constraints on both the range of peptides generated from a protein and the conformation in which they bind to MHC. As a result, the interaction of free peptides with MHC II can generate complexes that are distinct from those generated following antigen processing [51]. This influences which T cell epitopes induce tolerance. For example, a known peptide epitope from MBP was shown to bind to MHC in a dominant cryptic conformation when given in soluble form and failed to induce tolerance among self-antigen reactive T cells [52]. There are two important implications of this study; first, it shows that peptide epitopes generated by antigen processing are not guaranteed to induce tolerance; second, the results imply that tolerance induction with soluble peptides involves binding to MHC II without antigen processing. As a result, tolerogenic epitopes should be designed as antigen processing independent epitopes or apitopes so as to ensure that they bind MHC II in the correct conformation.
Santambrogio and colleagues have shown that immature DCs differ from other APC subsets, including B cells, monocytes and mature DC, by having ‘empty’ MHC II at the cell surface [53, 54]. This explains our recent results showing that tolerogenic peptides/apitopes selectively bind MHC on these steady-state/immature DC rather than B cells or monocytes when injected in soluble form in vivo (Shepard and Wraith, unpublished data). Peptides designed to mimic naturally processed epitopes without antigen processing selectively bind to tolerogenic DC and this explains why they induce tolerance. Repeated injection of these peptides in a dose escalation protocol promotes the generation of Tr1-like cells capable of preventing and controlling autoimmune diseases [38••].
Further work has indicated that the consequences of peptide treatment depend on ‘strength of signal’ as governed by dose and the MHC binding affinity of a given T cell epitope. While low to moderate affinity peptides induce T cell anergy, higher affinity analogues promote IL-10 secretion by the anergic T cells [55]. It is important to optimise peptide-MHC binding affinity given the critical role of IL-10 in bystander suppression and the anti-inflammatory properties of this cytokine.
Clinical experience with tolerogenic peptides
Antigenic peptides have been used to treat various hypersensitivity conditions including allergies [56, 57••], food hypersensitivity [58••] and autoimmune diseases including type 1 diabetes [59, 60••] and multiple sclerosis [61, 62, 63••]. These early phase clinical trials of immunotherapy with synthetic peptides provide supporting evidence that the mechanisms delineated in murine models translate well to man. Two overriding conclusions can be drawn: first, effective control of disease requires repeated exposure to the peptide antigens and second, that antigen-specific immunotherapy with synthetic peptides has been shown to be safe and well tolerated thereby allowing the repeated administration of peptide that will be required for effective control of autoimmune diseases.
Conclusions
This review has discussed various approaches used currently to induce antigen-specific immunotherapy of autoimmune diseases. This reveals how important it is to target tolerogenic DC with either free peptide or antigen associated with a carrier and/or immunosuppressive drug. The various approaches engage different mechanisms of tolerance (T cell deletion, anergy, induction of Foxp3+Treg or Tr1-like Treg cells): it is critically important to test these for efficacy in different autoimmune pathologies. Moreover, questions remain about the long-term safety of carrier materials when administered to patients repeatedly. At this time, only the soluble peptide approach has undergone extensive clinical testing and this, the most straightforward of the approaches reviewed, has proven safe and well tolerated.
Conflict of interest
David Wraith serves as Chief Scientific Officer for Apitope Technology (Bristol) Ltd and Apitope International NV on a consultative basis; is on the scientific advisory board for Apitope Intl NV and has sat on scientific advisory boards for Actelion Pharma, and Zealand Pharma; received travel funding from Apitope Intl NV; is a senior editor for Immunotherapy; holds patents for peptides, tolerisation-inducing composition, FVIII peptides and their use in tolerising haemophiliacs, composition, disease markers, tolerogenic peptides from myelin basic protein, peptide selection method, and improvements relating to influenza vaccine; has consulted for Peptide Therapeutics Ltd., Teva, GSK Bio, Hoffman La Roche, Novartis, DTI, and the Food Standards Agency; holds stock and stock options with Apitope Int. NV.