1. Introduction
Polymer science, regarding its subject of study, belongs to the oldest fields of science (existing since the times when living cells appeared), while as separate, well defined science it was formulated less than 100 years ago – between 1920 and 1930 – thanks to the pioneering works of the German chemist H. Staudinger. Another peculiarity of polymer science is that polymer materials have always been used by the human beings without having any idea what differs these materials from the others.
Nowadays, when to the natural polymer materials are added numerous synthetic ones with unknown in former time combination of properties, we can hardly imagine our life without these materials. What is more, their number and variety increases continuously because of the large opportunities offered by the new synthetic paths and the property of macromolecules – their chain character. This makes possible the preparation of completely new materials with desired properties or to drastically improve a property in a selected direction of already known material. In parallel to the many advantages of synthetic polymer materials, making them that important and attractive, they have also serious disadvantages as, for example, their adverse environmental impact.
The negative environmental impact of synthetic, petroleum-based polymers is possibly the main problem in polymer science and technology since this concern is increasing with the growth of plastics production and because of lack of acceptable solutions. The situation is getting even more serious if one takes into account the fact that polymer materials cannot compete with respect of their mechanical performance the other natural materials and, for this reason, they need to be reinforced. The most common reinforcing materials are glass- and carbon fibers, which cannot be subjected to incineration similarly to polymer matrix. Their amount is typically 1/3 (by weight) of the composite material.
Attempts for replacing glass and other inorganic fibers as reinforcing component for polymer composites resulted in application of natural fibers as reinforcement (e.g. Ref. [1]). The next step in this direction was the use of synthetic polymers as reinforcement but prepared as fibers, micro- or nanofibrils (e.g. Ref. [2]). Of course, this second approach is not as advantageous as using natural fibers that are biodegradable and eco-friendly. At the same time, the synthetic polymer–polymer composites seem to be much more acceptable from the environmental point of view in contrast to their inorganic counterparts because they, being organic in nature, can be incinerated.
The second important problem of polymer science and technology is related with their mechanical properties, even in case of reinforced polymers. It is related with the known fact that the strength of the reinforcement is not fully utilized as the matrix and reinforcement do not have a perfect adhesion. This is because the two basic components of the composite material have different chemical composition. Creating polymer–polymer composites is one possible partial solution as polymers are of course much closer chemically to each other than their mineral counterparts, and so easier to choose for good adhesion. For example, while in the polymer–polymer composites both component are hydrophobic, in the glass fiber reinforced polymer composites the matrix is hydrophobic and the reinforcement – hydrophilic.
Another partial solution of this problem is found in the use of a third component having in its chemical composition something in common with the matrix as well as with the reinforcement. It is situated on the phase boundary between the two components and thus reducing the drastic difference between them. Good example of these compounds is the use of compatibilizers.
A situation of ideal adhesion could be observed when between the matrix and the reinforcement chemical bonds are established. This can happen if the matrix and the reinforcement belong to the group of condensation polymers(e.g. polyesters and polyamides) or contain functional groups (e.g. cellulose) capable to react with each other. In the first case are possible transreactions:
And in both cases – additional condensation can take place:
It should be noted that formation of covalent chemical bonds on the interface by far does not exhaust the possibilities for chemical interaction of partners in polymer blends and composites – hydrogen bonding is the next opportunity.
In author's opinion this potential for drastic improvement of the interfacial adhesion quality between matrix and reinforcement is not exploited enough by the composite community possibly because of lack of chemistry knowledge among the engineers involved in this type of research.
Another case of ideal adhesion between matrix and reinforcement can be realized if the two basic composite components, the matrix and the reinforcement, are of the same chemical composition. This is the case of single polymer composites.
The concept of single polymer composites was formulated some 40 years ago by Roger Porter and demonstrated in his publication with N. Capiati [3]. They used two types of samples of high density polyethylene (HDPE) differing mostly in their melting temperatures. The preparation of this new material, called by them “one polymer composite” was possible due to the fact that aligned and extended chains provide thermodynamically more stable crystals, which thus will have higher melting points than conventionally crystallized melts.
During the last two decades the interest of academia and industry in single polymer composites (SPCs) increased immensely due to the environmental impact of synthetic polymers and their glass fiber reinforced composites.
Fundamental contributions to the field of single polymer composites were made by Ward and his co-workers (e.g. Refs. [4], [5], [6], [7], [8], [9], [10], [11], [12]). They not only succeeded to prepare SPCs of large number of homopolymers, to develop new techniques for their manufacturing but they also patented [8] and commercialized products with SPC structure. Interesting contribution to SPCs was made also by J. Karger Kocsis (e.g. Ref. [13]) demonstrating that such composites can be prepared using the polymorphic phenomenon in polymers. And, finally, to the SPCs was recently added the family of nanofibrillar single polymer composites prepared from parallel aligned non-interconnected nanofibrils (diameters between 50 and 250 nm) having superior mechanical properties (e.g. Refs. [14], [15]).
The single polymer composites will remain in the focus of interest further on because of their advantages: (i) lack of matrix-reinforcement adhesion problem, (ii) high aspect ratio (always a fibrous constituent is involved), (iii) strongly dominating reinforcing constituent (up to 80 or 90%), (iv) environmentally friendly (no mineral additives), (v) complete regeneration of SPCs, (vi) for the preparation of nanofibrillar SPCs a nano-size material only is used, and (vii) the nanofibrillar SPCs demonstrate extremely high mechanical properties.
This prediction is based on the fact that they are lightweight (their density is lower than most of the traditional composites), environmentally benign (especially due to their easy recycling via reprocessing in the melt), and they offer novel properties (related to nanoporosity, for example). For this reason they enjoy a continuously increasing interest as demonstrated by the very recent reviews on this topic [13], [14], [15], [16], [17], [18], [19]. To their basic disadvantages belongs the very narrow processing window – in many cases a couple of degrees °C only.
And finally, in order to be able to make use of the unique properties of single polymer composites outlined above we should not forget that they are one-component systems where the matrix and the reinforcement are of the same chemical composition. They must not be mixed up with the closely related to them polymer–polymer composites where the matrix and the reinforcement have different chemical composition.
The target of this review paper is to demonstrate that the catastrophic difference between the expected mechanical properties and the observed ones of polymer nanocomposites prepared by melt blending of matrix and reinforcement is related with the poor dispersion resulting in formation of micro- instead of nanocomposites. A second target of the review is to offer method for nanocomposite preparation free of the dispersion step so far techniques and instrumentation for a proper dispersion of reinforcement are missing.
2. Polymer nanocomposites – preparation, mechanical performance, problems
About three decades ago started the preparation of nanomaterials. According to the recent definition of the European Commission [20] 50% or more of the particles of these materials in their number size distribution is in the size range 1–100 nm in at least one dimension.
All nano-sized materials, regardless of their chemical composition and method of manufacturing, have a common characteristic feature – the extremely high ratio of surface area to volume. For example, 1 kg of particles of 1 mm3 has the same surface area as 1 mg of particles of 1 nm3. The natural tendency to reduce this free surface is the driving force for agglomeration of nanoparticles in larger formations.
Discovery with a paramount importance for the polymer science was the synthesis of carbon nanotubes, usually credited to Iijima [21]. It turned out that carbon nanotubes are the strongest material ever created – with an elastic modulus in the terapascal range carbon nanotubes (CNTs) overcome any known material. The idea to use CNTs as reinforcement of plastics arose immediately because simple model calculations demonstrated that small amounts of this reinforcement will result in some 10-fold increase of the elastic modulus.
The idea was accepted very well with the expectation that the basic, the environmental problem of polymer science and technology, will be perfectly solved by replacement of the common 30% (by wt.) glass or carbon fibers with 1–2% (by wt.) nano-size material and getting at the same time at least 10-times improvement of elastic modulus! In addition, the preparation of these new polymer nanocomposites can be performed on the common equipment for polymer processing via melt blending of polymer matrix with one of the many nano-size inorganic materials. Some 1000 papers on polymer nanocomposites have been published annually following this scenario.
What about the mechanical properties of these polymer nanocomposites? The numerous publications on polymer nanocomposites demonstrated that their mechanical performance is rather modest – improvements (if any!) of elastic modulus by 20% and the tensile strength by 50% are typical [22], [23], [24]. Only in exceptional cases the improvement is larger than 30% [23].
This curious situation was summarized some 10 years ago by Schaefer et al. in their review “How Nano Are Nanocomposites?” [25]: “Composite materials loaded with nanometer-sized reinforcing fillers are widely believed to have the potential to push polymer mechanical properties to extreme values. Realization of anticipated properties, however, has proven elusive. With the exception of reinforced elastomers, nanocomposites have not lived up to expectations. Although claims of modulus enhancement by factors of 10 exist, these claims are offset by measurements…” [25].
Unfortunately, the situation did not change even nowadays as follows from the statement in a very recent (2017) review on the practical applicability of polymer nanocomposites [26]: “Initial developments of nanocomposite thermosets focused on high aspect ratio nanoparticles such as nanoclays, carbon nanotubes and more recently graphenes. Generally, these systems showed 10 – 35% improvement in mechanical properties with 0.2 – 5 wt. % filler. However, the translation of these improvements to prepregs or laminates proved to be difficult due to processing issues, including extremely high viscosity, nanoparticles filtration, nanoparticles agglomeration, and void formation” [26].
The extremely large discrepancy between the expectations and experimentally observed mechanical results of polymer nanocomposites prepared via blending the two basic composite components is obvious. The important question regarding the reasons for this drastic discrepancy arises.
The concept of polymer nanocomposites failed for the following reasons: (i) poor dispersion, (ii) poor interfacial load transfer, (iii) process-related deficiencies, (iv) poor alignment, (v) poor load transfer to the interior of filler bundles, (vi) the fractal nature of filler clusters. Among all these factors the very first one seems to be the most important also because it helps us to correctly define the type of composite material – macro-, micro-, or nanocomposite, depending on the degree of dispersion, i.e. depending on the sizes of the single reinforcing particles.
It seems interesting to try to answer the question when we are dealing with nanocomposites. In the case of common composites (macrocomposites), e.g. glass- or carbon fibers reinforced ones, as well as in the case of microcomposites(where the reinforcing particles have sizes in micrometer range), each individual reinforcing element is surrounded by matrix material. Dealing with nanocomposites one should expect the same situation, i.e. each single nanoparticle must be individually surrounded by matrix. This means that no aggregation of the nanoparticles must take place and the dispersion process should go to single individual nanoparticles. With respect of the degree of dispersion, this process is very alike to the preparation of true solutions, i.e. solutions in which the dispersion goes to single molecules.
How does look the situation about the dispersion degree in the case of polymer nanocomposites? Unfortunately, this problem is seriously underestimated by all the researchers. As a rule, in each of thousands publications on polymer nanocomposites one can see one or two electron microscopic photographs of single nanoparticles but the dispersion character of particles practically is not discussed. In the best case it is mentioned that aggregates of nanoparticles can also be observed. But that these aggregates dominates, that they comprise many thousands or millions of nanoparticles (aggregate sizes are in the micrometer range while the nanoparticles of inorganic materials are typically 2–5 nm!) usually it is not mentioned. The authors of such publications incorporate in the title the word “nanocomposite” only because they blended a polymer with a nano-size material. Practically, no serious attempts to analyse the degree of dispersion of the reinforcement into the matrix are undertaken.
Let remind here that the electron microscopy offers a good idea regarding the shape and sizes of the observed particles – single or aggregated ones in this particular case, and, as a matter of fact, no information regarding the total amount of objects with a given size. The scattering techniques are doing just the opposite job. On this detail it is also stressed in the above mentioned recent review [26]. For the case of polymer nanocomposites useful application could have the light scattering.
From the physics it is known that capable to scatter are only particles whose sizes are comparable with the size of the wave length of the applied light. Luckily, such studies are available. Of particular interest are the polymer nanocomposites where the matrix represents an inherently transparent material, as for example the atactic poly(methyl methacrylate) (at-PMMA) and the atactic polystyrene (at-PS). Very close to the group of atactic polymers are the polycarbonates (PC) and polyarylates (PAr) because of their poor crystallization ability.
The neat at-PMMA and at-PS are completely transparent materials (the transmission of the light is of 100%) and for this reason they are widely used for replacement of inorganic glasses. This situation changes drastically if to such polymers are added very small amounts (1–2 wt. %) of non-transparent nano-size material – the light transmission ability is reduced. Such data are summarized in Table 1.
Table 1. Examples of light transmission of polymer nanocomposites where the matrix is inherently transparent material (light transmission of 100%).
| # | Transmission of light | Specimen thickness | Filling degree | Matrix | Filler size | Ref. |
|---|---|---|---|---|---|---|
| Matrix of at-PMMA | ||||||
| 1 | 95% (600 nm, 20 wt. %) | Thin | 0–20 wt. % | PMMA | ZrO2/SiO2(<100 nm) | [27] |
| 2 | 2% (1 wt. %, 600 nm) | 3.5 mm | up to 1 wt. % | PMMA | ZnO (75 nm) | [28] |
| 50% (0.01 wt. %) | 0.01 wt. % | [28] | ||||
| <90% (0.01 wt. %, prepolymer) | 0.01 wt. % | [28] | ||||
| 3 | 80% (600 nm, 15 wt. %) | – | 0–15 wt. % | PMMA | ZrO2(4 nm) | [29] |
| 4 | 90% (600 nm) | 4 mm | 0.5 wt. % | PMMA | ZnO (2.3 nm) | [30] |
| 5 | >85%, above 400 nm | 0.127 mm | 0.26 wt. % | PMMA | SWNT | [31] |
| 6 | Increased absorb.>600 nm - | 100–300 μm | – | PMMA | Fe2O3 (8–200 nm) | [32] |
| Matrix of PS | ||||||
| 7 | Good, brown coloured, 550 nm | 1 mm | 3–15 wt. % | PS | Fe3O410 × 50 nm | [33] |
| 8 | “Quite” transparent, >550 nm | 100 μm | 5–15 wt. % | PS | Fe3O410 × 50 nm | [33] |
| 9 | Decreases to 75% (20 wt. %) | 2.5 μm | up to 95 wt. % | PS | CeO (20 nm) | [34] |
| 10 | High | 20 nm (LED) | Zn: 8.2, Cu: 0.12 wt. % | PS (Sulphonated) | ZnS:Cu (3 nm) | [35] |
| Matrix of other transparent polymers | ||||||
| 11 | 80% (2 wt. %, 600 nm) | 0.15 mm | 2 wt. % | PC | AlO2(whisker) | [36] |
| 12 | 52% (1 wt. %, 600 nm) | 2 mm | 1 or 2 wt % | PC | Al2O3(96 nm) | [37] |
| 13 | 87% (600 nm, 15 wt. %) | 0.12–0.29 mm | 0–15 wt. % | PAr | Al2O3 | [38] |
From the data presented in Table 1 one can conclude that: (i) the polymers used as matrix are inherently completely transparent materials, (ii) the reinforcing nanomaterials used have dimensions below 100 nm (typically a couple of nm for the majority of cases), and (iii) for the light transmission measurement a light with a wave length between 500 and 600 nm has be used. If the studied composites are really polymer nanocomposites, i.e. materials in which the reinforcing component is dispersed to single nanoparticles, these composites should have the same transparency as the neat matrices since the single particles are too small in order to scatter. The reduction in transparency (Table 1) means that the particles in the matrix are much larger, approaching the size of the wave length of the light used (500–600 nm), i.e. one deals practically with large aggregates of nanoparticles.
It appears that currently we do not have reliable tools and/or techniques for a proper dispersion of nanomaterials in polymer melts in order to reach the dispersion degree of single nanoparticles what is the requirement for having true nanocomposites. Obviously, as long as the respective tools and techniques are missing, for the preparation of such composites one must use methods free of the dispersion step in the manufacturing process.
3. Solution of the dispersion problem
Do dispersion-free methods for preparation of polymer nanocomposites exist? As a matter of fact, there are currently at least two techniques allowing avoiding the dispersion problem in preparation of polymer nanocomposites. Both of them result in true nanocomposites, i.e. when the nano-size reinforcing component reaches dispersion to single nanoparticles.
The two approaches are the essence of the relatively new “concept of converting instead of adding” [15]. This means that instead to take the two basic composite components, the matrix and the reinforcement, in their final form and blend them, one takes one component only and during the processing creates the missing second component. For example, starting from blend of two thermodynamically non-miscible polymers, we can convert the minor blend component into nanofibrils and thus to prepare a nanofibrillar polymer–polymer composite (PPC).
If we have neat polymer nanofibrils, by means of thermal treatment close but below the temperature of complete melting we can produce a small amount of isotropic matrix of the same polymer (playing the role of binder of nanofibrils) and in this way to prepare a nanofibrillar single polymer composite (SPC).
Both techniques are free of the dispersion step and, what is more important, one always observes a perfect distribution of the reinforcing nanomaterial in the polymer matrix, i.e. each nanofibril is individually surrounded by the matrix polymer and no aggregation is observed on the scanning electron microscopy (SEM) micrographs.
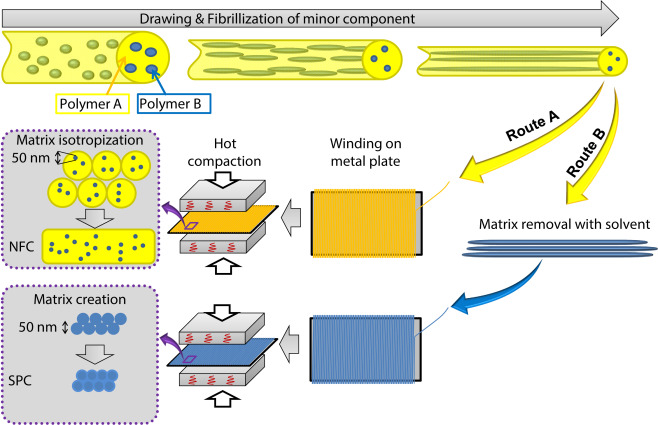
The manufacturing process of nanofibrillar PPCs and SPCs is schematically shown in Fig. 1. It should be noted that this process can be performed on common equipment for polymer processing. The two thermodynamically non-miscible polymers in a usual ratio A/B = 70/30 (by wt.) are melt blended, extruded and cold drawn. Thereafter, depending on the target, one goes the Route A or B (Fig. 1).
 Fig. 1. Manufacturing of nanofibrillar PPCs (NFC) (Route A) and nanofibrillar SPCs (Route B) via the “concept of converting instead of adding”.
Fig. 1. Manufacturing of nanofibrillar PPCs (NFC) (Route A) and nanofibrillar SPCs (Route B) via the “concept of converting instead of adding”.If nanofibrillar polymer–polymer composite is needed, the drawn bristle is winded on a metal plate and subjected to compression moulding at melting temperature of the matrix polymer A, which has to be at least 40 °C below the melting of the reinforcing polymer B (Fig. 1, Route A).
For preparation of nanofibrillar single polymer composites one has to select the Route B (Fig. 1), according to which from the drawn bristles has to be removed the matrix polymer A using a selective solvent. The rest of nanofibrillar bunch of B has to be winded on a metal plate and compression moulded at temperature at least 20 °C below the melting temperature of B (Fig. 1, Route B). In this way, due to a partial surface premelting, a small amount of isotropic matrix (binder of nanofibrils) is created (Fig. 1, Route B).
If we analyse the prepared two materials, the PPC and the SPC, by means of SEM, we can get important information regarding the sizes of the reinforcing nanofibrils and, what is more interesting in the current case, the distribution of the reinforcing nanofibrils in the matrix. Such results for the two materials under discussion are shown in Fig. 2.
 Fig. 2. SEM micrographs of polyethylene/poly(vinylidene fluoride) (PE/PVDF = 70/30 by wt.) nanofibrillar polymer–polymer composite (a and b) and PET nanofibrillar single polymer composite (c and d): a) and c) – cryofracture perpendicular to the nanofibril orientation, b) – cryofracture parallel to the nanofibrils orientation, and d) – the surface of the SPC film shown in c).
Fig. 2. SEM micrographs of polyethylene/poly(vinylidene fluoride) (PE/PVDF = 70/30 by wt.) nanofibrillar polymer–polymer composite (a and b) and PET nanofibrillar single polymer composite (c and d): a) and c) – cryofracture perpendicular to the nanofibril orientation, b) – cryofracture parallel to the nanofibrils orientation, and d) – the surface of the SPC film shown in c).The photographs taken from the cryofractures perpendicular to the nanofibrils orientation (Fig. 2a and c, as well as from cryofracture parallel to the nanofibrils orientation (Fig. 2b) demonstrate rather homogeneous distribution of the reinforcing nanofibrils in the polymer matrix, i.e. practically, no aggregates of nanofibrils can be observed. Such a situation is quite different from the case of polymer nanocomposites prepared via blending of the two starting components as demonstrated in the previous paragraphs.
Coming back to the preparation of these true nanofibrillar polymer composites it should be stressed that the formation of the reinforcing nanofibrils takes place during the melt blending followed by extrusion and cold drawing as schematically shown in Fig. 3.