1. Introduction
Metabolic engineering is enjoying an auspicious moment, when its potential is becoming evident in the form of many commercially available products with undeniable impact on society. This discipline has produced: synthetic silk for clothing (Hahn, 2019; Johansson et al., 2014), meatless burgers that taste like meat because of bioengineered heme (“Meat-free outsells beef,” 2019), synthetic human collagen for cosmetic purposes ("Geltor unveils first biodesigned human collagen for skincare market", 2019), antimalarial and anticancer drugs (Ajikumar et al., 2010; Paddon and Keasling, 2014), the fragance of recovered extinct flowers (Kiedaisch, 2019), biofuels (Hanson, 2013; Peralta-Yahya et al., 2012), hoppy flavored beer produced without hops (Denby et al., 2018), and synthetic cannabinoids (Dolgin, 2019; Luo et al., 2019), among others. Since the number of possible metabolites is enormous, we can only expect these successes to significantly increase in number in the future.
Traditional approaches, however, limit metabolic engineering to the usual 5–15 gene pathway, whereas full genome-scale engineering holds the promise of much more ambitious and rigorous biodesign of organisms. Genome-scale engineering involves multiplex DNA editing that is not limited to a single gene or pathway, but targets the full genome (Bao et al., 2018; Esvelt and Wang, 2013; Garst et al., 2017; Liu et al., 2015; Si et al., 2017). This approach can open the field of metabolic engineering to stunning new possibilities: engineering of microbiomes for therapeutic or bioremediation uses (Lawson et al., 2019), designing of multicellular organisms as biomaterials that match a specification (Islam et al., 2017), ecosystem engineering (Hastings et al., 2007), and perhaps even fusion of physical and biological systems. None of these examples are likely to become reality through a traditional trial-and-error approach: the number of genetic part combinations that could produce these outcomes is a vanishingly small fraction of the total possible. For example, engineering a microbiome to produce a medical drug involves not only introducing and balancing the corresponding pathway in one or more of the microbiome species, but also modifying internal regulatory networks so as to keep the community stable and robust to external perturbations. Even for the case of single pathways and teams of highly-trained experts, the trial-and-error approach is hardly sustainable, since it results in very long development times: for example, it took Amyris an estimated 150 person-years of effort to produce the immediate precursor of the antimalarial artemisinin, and Dupont 575 person-years to generate propanediol (Hodgman and Jewett, 2012). An approach that pinpoints the designs that match a desired specification is needed.
The main challenge in more sophisticated biodesign is, arguably, our inability to accurately predict the outcomes of bioengineering (Carbonell et al., 2019; Lopatkin and Collins, 2020). New technologies provide markedly easier ways to make the desired DNA changes, but the final result on cell behavior is usually unpredictable (Gardner, 2013). If metabolic engineering is “the science of rewiring the metabolism of cells to enhance production of native metabolites or to endow cells with the ability to produce new products” (Nielsen and Keasling, 2016), the ability to engineer a cell to a specification (e.g. a given titer, rate and yield of a desired product) is critical for this purpose. Only the ability to accurately predict the performance of a genetic design can avoid an arduous trial-and-error approach to reach that specification.
Moreover, while the flourishing offshoots of the genomic revolution provide powerful new capabilities to discover new DNA sequences, understand their function, and modify them, it is not trivial to harness these technologies productively. The genomic revolution has provided the DNA code as a condensed set of cell instructions that constitutes the main engineering target, and functional genomics to understand the cell behavior. Furthermore, the cost for these data is rapidly decreasing: sequencing cost decreases faster than Moore's law, transcriptomic data grow exponentially (Stephens et al., 2015), and high-throughput workflows for proteomics and metabolomics are slowly becoming a reality (Chen et al., 2019; Zampieri et al., 2017). But many researchers find themselves buried in this “deluge of data”: there seems to be more data than time to analyze them. Furthermore, data come in many different types (genomics, transcriptomics, proteomics, metabolomics, protein interaction maps, etc), complicating their analysis. As a result, analysis of functional genomics data often does not yield sufficient insights to infer actionable strategies to manipulate DNA for a desired phenotype. Moreover, CRISPR-based tools (Doudna and Charpentier, 2014; Knott and Doudna, 2018) provide easy DNA editing and metabolic perturbations (e.g. CRISPRi (Tian et al., 2019)). These tools provide the potential to perform genome-wide manipulations in model systems (Wang et al., 2018), and a growing number of hosts (Peters et al., 2019). However, it is not clear how to prioritize the possible targets. Rational engineering approaches have proven useful in the past (George et al., 2015; Kang et al., 2019; Tian et al., 2019), but the detailed knowledge of a pathway can produce on the order of tens of targets, whereas CRISPR-based tools can reach tens of thousands of genome sites (Bao et al., 2018; Bassalo et al., 2018; Garst et al., 2017; Gilbert et al., 2014).
Machine learning (ML) is a possible solution to these problems. Machine learning can systematically provide predictions and recommendations for the next steps to be implemented through CRISPR (or other methods (Paschon et al., 2019; Reyon et al., 2012; Wang et al., 2019)), and it can use the exponentially growing amounts of functional genomics data to systematically improve its performance. Machine learning has already proven its utility in many other fields: self-driving cars (Duarte and Ratti, 2018), automated translation (Wu et al., 2016) , face recognition (Voulodimos et al., 2018), natural language parsing (Kreimeyer et al., 2017), tumor detection (Paeng et al., 2016), and explicit content detection in music lyrics (Chin et al., 2018), among others. It has the potential to produce similar breakthroughs in metabolic engineering.
However, a change in perspective is required regarding the relative importance of molecular mechanisms. Whereas the machine learning paradigm concentrates on enabling predictive power, metabolic engineers typically define scientific value around the understanding of genetic or molecular mechanisms (see section 4.0). Nonetheless, the biological sciences (including computational biology) have been particularly challenged to make accurate quantitative predictions of complex systems from known and tested mechanisms. Hence, if accurate quantitative predictions are needed for a more transformative metabolic engineering, it may be desirable to shift some of the emphasis from identifying molecular mechanisms into enabling data-driven approaches. This apparent detour may, in the end, more efficiently produce mechanistic models, if we combine the predictive power of machine learning with the insight of molecular mechanisms (Heo and Feig, 2020).
In this review we provide an explanation of machine learning in metabolic engineering terms, in the hopes of providing a bridge between both disciplines. We explore the promises of machine learning, as well as its current pitfalls, provide examples of how it has been used so far, as well as auspicious future uses. In short, we will make the case that machine learning can take metabolic engineering to the next step in its maturation as a discipline, but it requires a conscious choice to understand its limitations and potential.
2. Demystifying machine learning for bioengineers
2.1. What is machine learning?
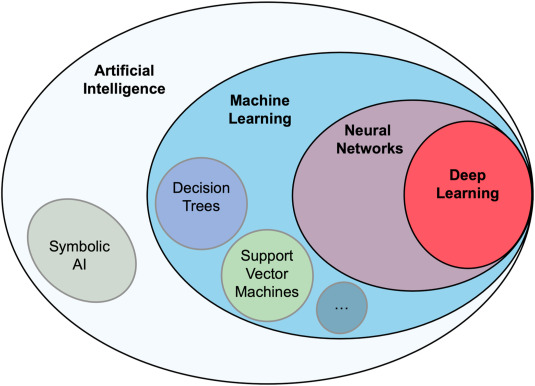
Machine learning is a subdiscipline of Artificial Intelligence (AI), which attempts to emulate how a human brain understands, and interacts with, the world (Fig. 1). A fully functioning AI would enable us to perform the same processes as human metabolic engineers: choose the best molecules to produce, suggest possible pathways to produce it, select the right pathway design to obtain the desired titers, rates and yield, and interpret the resulting experimental data to troubleshoot the metabolic engineering effort. A fully functioning AI would of course be useful for many other tasks such as: fully autonomous cars and planes, recommending medical treatments, directing agricultural practices, reading and summarizing texts like a human, automating translations from different human languages, and producing music and movies. Obviously, we do not yet have full functioning AIs (or strong AI or artificial general intelligence as it is often referred to (Pei et al., 2019; Walch, 2019 )), and it is a continuing debate whether we will ever have them (Melnyk, 1996), but AI approaches have been quite successful in some bounded tasks such as playing chess and Go better than humans (Silver et al, 2016, 2018), or predicting protein structures from sequence (AlQuraishi, 2019). Since AI and machine learning are generally applicable tools, some of these partial successes can be very useful for metabolic engineering (see section 3 for examples).
 Fig. 1
Fig. 1Machine learning is the study of computer algorithms that seek to improve automatically through experience (i.e. learning), often by training on supervised examples (Fig. 2), also known as supervised machine learning. This works by statistically linking an input to its associated response for several different examples: e.g. promoter choice for a pathway and the corresponding final production, protein sequence and its function, etc (Fig. 2, Fig. 3). It is important to realize that the emphasis is set in predicting the response, rather than produce mechanistic understanding. In fact, the algorithm linking input and response is not meant to represent a mechanistic understanding of the underlying processes: for example, modeling the full process of promoters causing the expression of proteins that code enzymes which then catalyze reactions that transform metabolites and result in a predicted production. Rather, the algorithm is chosen to be as expressive as possible to be able to learn any relationship between input and response. Hence, none of the biological information is encoded in the algorithm; all the biological information is provided by the training data, which must be carefully selected (supervised) so the algorithm can learn the desired relationship (promoters to production, protein sequence to function, etc.), generalize it, and be able to predict it for new inputs that were not in the training set (Fig. 3). This difference is crucial with respect to traditional metabolic engineering and microbiology, where understanding the mechanism is considered of paramount importance (see section 2.2.1 for a specific example). In machine learning, we can see the situation in which we can predict that, e.g., a given promoter choice will have the best production, but we cannot explain the metabolic mechanism that provides that optimal production (Zhang et al., 2020). This state of affairs has its pros and cons, and efforts have been made to introduce biological prior knowledge in the algorithms (see section 4).
 Fig. 2
Fig. 2 Fig. 3
Fig. 3 Fig. 4
Fig. 4 Fig. 5
Fig. 5 Fig. 6
Fig. 6 Fig. 7
Fig. 7There is a continuous interplay between the complexity of a supervised machine learning algorithm and the amount of data available to train it (Fig. 4). If the model/algorithm is not expressive enough (not enough parameters), it will be unable to describe the data accurately (underfitting). If the model displays much more parameters than data instances are available, it will just “memorize” the training data set rather than grasp the underlying general patterns required to predict new inputs (overfitting). In this case, the algorithm will produce exceedingly good results for the training set, but very poor ones for any new input that is used as a test (Fig. 3, Fig. 4). Cross validation (Fig. 3) provides an effective way to choose the number of parameters: both overfitting and underfitting result in very poor predictions.
There are many supervised machine learning algorithms available in the public domain: linear regressions, quadratic regressions, random forest, support vector machines, neural networks, Gaussian process regressors, gradient boosting regressors (the popular library scikit-learn provides a good starting point with an extensive list and explanations (Pedregosa et al., 2011)). To give a concrete example, a classic machine learning algorithm is the decision tree, that can be used, for example, to predict which protein expression levels result in high production (Fig. 5). As can be observed, this algorithm represents a high-level abstraction of how humans are believed to think. Because no single algorithm is best for every learning task (Wolpert, 1996), a significant endeavor when applying machine learning is choosing the optimal algorithm for your problem (and its hyperparameters, see Fig. 5). Ensemble modeling is an alternative approach that sidesteps the challenge of model selection (Radivojević et al., 2020). Ensemble modeling takes the input of various different models and has them “vote” for a particular prediction. Based on their performance, a different weight is assigned to each algorithm. The examples of the random forest algorithm (Ho, 1995) or the super learner algorithm (van der Laan et al., 2007) have demonstrated that even very simple models can increase their performance significantly by using an ensemble of them (e.g., several decision trees in a random forest algorithm).
Learning without supervision also constitutes an important part of machine learning, given the significant effort involved in creating labeled data sets. The areas of machine learning focused on this challenge are unsupervised learning and reinforcement learning. Unsupervised learning searches for patterns in a data set with no pre-existing labels, requires only minimal human supervision, and often attempts to create clusterings or representations that aid human understanding or reduce dimensionality (Fig. 6). Examples of unsupervised machine learning algorithms include Principal Component Analysis (PCA), K-means clustering (Sculley, 2010), and Single Value Decomposition (Manning et al., 2008). Familiar examples in metabolic engineering include identifying patterns in metabolomics profiles that distinguish between different types of cells: healthy vs. sick (Sajda, 2006), stressed vs. non-stressed (Luque de Castro and Priego-Capote, 2018; Mamas et al., 2011), or high-producing vs low-producing (Alonso-Gutierrez et al., 2015). Reinforcement learning represents a different paradigm regarding learning from experience that posits that humans learn not from properly labeled examples, but rather from interacting and probing their environment. Hence, the aim of reinforcement learning is to use experience and data to update an internal policy that optimizes a desired goal (Fig. 6). A prime example of this approach (Treloar et al., 2020) is controlling a bioreactor which contains a co-culture (environment), through manipulations of the concentration of auxotrophic nutrients flowing into the reactor (actions), and informed by the relative abundances (measurements), to ensure a specified co-culture composition (goal). Perhaps the most known example of reinforcement learning are the Hidden Markov Models (HMMs) that are commonly used to annotate genes and align sequences (Yoon, 2009). Reinforcement learning has also been applied to suggest pathways for specific molecules (Koch et al., 2020) or molecules that fit desired properties (Popova et al., 2018), as well as to optimize large-scale bioreactor fermentations using online continuous process data (see section 3.3). However, there is still generally a dearth of reinforcement learning examples in metabolic engineering, which represents an opportunity for this type of machine learning.
Deep learning (DL, Fig. 7 (LeCun et al., 2015)) is a specific type of machine learning algorithm that has been particularly successful in the past decade (Fig. 8). This algorithm has been shown to improve performance with the amount of training data when other methods plateau. Deep learning is based in Artificial Neural Networks, which attempt to mimic how neurons work (Fig. 7). In the last decade, deep learning has been the basis of the most celebrated AI achievements. However, compared to more classical machine learning methods, deep learning generally requires far larger amounts of data for training: 10,000s or millions of instances, as opposed to hundreds or thousands (although that depends on the number of inputs, see section 2.2.2). The reason for these large data sets hunger is that deep neural networks can include thousands to millions of parameters, which need to be determined from the data (see Fig. 4). In metabolic engineering, the use of deep learning has been sparse for this reason: the data sets tend to be small (<100 instances), with the notable exception of sequence data. Deep learning has been most useful with sequence data: e.g., to predict protein function (Ryu et al., 2019), or translation initiation sites (Clauwaert et al., 2019) (see section 3.0). However, this is expected to change as more high-throughput methods to characterize cellular components become available, provided that data are structured consistently and stored appropriately (see section 2.4). Indeed, techniques to generate high quality omics data are improving rapidly and the cost per sample is decreasing (Stephens et al., 2015), so application of deep learning to metabolic engineering might become commonplace soon.
2.2. A couple of illustrative examples of machine learning in metabolic engineering
We will now illustrate how machine learning algorithms work through two different applications that elucidate particularly important points: predicting the kinetics of a metabolic network, and optimizing cell-free butanol production. We have focused on these examples because we believe they most relate to the day-to-day activities of metabolic engineers: leveraging omics data and improving production.
2.2.1. Kinetic learning: relearning Michaelis-Menten dynamics through machine learning
Our first example uses machine learning to tackle a commonly encountered problem in bioengineering: predicting the kinetics of a metabolic pathway. Predicting pathway dynamics can enable a much more efficient pathway design by allowing us to foresee in advance which pathway designs will meet our specifications (e.g., titers, rates and yields). Classic kinetic models predict the rate of change of a given metabolite based on an explicit functional relationship between substrate/product concentrations (metabolites) and enzymes (protein abundance, substrate affinity, maximum substrate turnover rate). Michaelis-Menten kinetic models (Costa et al., 2010; Heinrich and Schuster, 1996) have historically been the most common choice. In reality, the true functional relationship between metabolites and enzymes are typically unknown for most reactions due to gaps in our understanding of the mechanisms involved, resulting in poor prediction capabilities.
Costello et al. (Costello and Martin, 2018) showed that supervised machine learning (Fig. 2) can offer an alternative approach, where the relationship between metabolites and enzymes can be directly “learnt” from time series of protein and metabolite concentration data. In a sense, this approach involves relearning the equivalent of Michaelis-Menten based purely on data. This is a prime example of how a machine learning approach ignores mechanism in favor of predicting power: there is no intention that the function predicting metabolite change rate from proteins and metabolites describes a mechanism, but it offers the best prediction of the final limonene/isopentenol, which is what we require for our engineering. In this case, the inputs (Fig. 3) were the exogenous pathway protein and metabolite concentrations, and the response was the rate of change of the metabolite. The instances involved each of the time points for which the metabolite rate of changes was learnt.
This approach outperformed a classic kinetic model in predictive power using very little data: only three time series of protein and metabolite measurements of 7 time points each (for two different pathways). While it would be desirable to have hundreds of time point measurements, the high cost and time associated with performing multi-omic experiments typically constrains data sets to less than 10 time points/samples, which is too sparse for training accurate models. Critical to its success, hence, was the use of data augmentation to increase the number of available instances from the initial 7 time points to the final 200 used for learning. Data augmentation simulates additional instances by modifying or interpolating actual data. In this case, data augmentation involved first smoothing the data (via a Savitzky-Golay filter) and then interpolating new data points from the fitted curve. This augmentation scheme only assumed continuity and smoothness between time points, but provided sufficient data to train a machine learning model using data from only 2 time series that accurately predict pathway dynamics of the “unseen” third strain. The final predictions of metabolite concentrations for the exogenous pathways, although not perfect by any measure, were more accurate than equivalent predictions by a hand-crafted kinetic model. More importantly, while the kinetic model took weeks to produce through arduous literature search, the kinetic learning approach can be systematically applied to any pathway, product and host with no extra overhead.
An opportunity to improve the machine learning model predictions of Costello et al. would of course be to collect more data, but deciding which data to collect is not always clear. For example, instead of using protein and metabolite data only from the exogenous pathways as input features, protein and metabolite measurements from the full host metabolism could be added (surely, host metabolic effects like ATP supply must be relevant). However, using these extra data would not necessarily improve machine learning predictions. This is because many machine learning algorithms suffer from the “curse of dimensionality”: that is, the amount of data needed to support results in a statistically sound fashion often grows exponentially with the dimensionality of the input (Fig. 4). Hence, machine learning algorithms may struggle to learn from data sets that have many measurements or “input features” (columns), but few instances (rows). Adding host proteins and metabolites will increase the number of inputs without increasing the number of instances. Unfortunately, most multi-omic data sets used in metabolic engineering fit this description, containing more than 5000 measurements (e.g. proteins or metabolites abundances), but only tens to hundreds of instances (e.g. different time points, strains, or growth conditions, depending on what your algorithm is attempting to learn) (Fig. 4). Therefore, collecting as many instances as possible should be emphasized early on during experimental design (see section 2.4).
In the absence of being able to generate more data, algorithms that reduce the number of input features to the most important ones can be performed, a process known as feature selection. Feature selection (Pedregosa et al., 2011) was used in Costello et al. (Costello and Martin, 2018) to identify a subset of the input features based on their contribution to the model's error. This, more limited, curated set of features was then used to predict metabolite dynamics. The idea behind this is to remove non-informative or redundant input features from the model. An additional approach used was dimensionality reduction, where “synthetic features” are created that transform the original input features into fewer ones (or “lower dimensions”) based on their contribution to explaining the data's variability (for example, via principal component analysis). Similar to feature selection, these algorithms simplify the data set in order to better fit a machine learning model. These approaches were integrated into a machine learning pipeline using the tree-based pipeline optimization tool (TPOT) (Olson et al., 2016; Olson and Moore, 2019), which automatically selected the best combination of feature preprocessing steps and machine learning models from the scikit-learn library (Pedregosa et al., 2011) to maximize prediction performance.
2.2.2. Artificial neural networks to improve butanol production in cell-free systems
Our second example involves using deep neural networks to optimize cell-free butanol production (Karim et al., 2020). Here, the authors provide an example of how machine learning can accelerate the design-build-test-learn (DBTL) cycles used in metabolic engineering (Nielsen and Keasling, 2016), by effectively guiding pathway design. In this study, the authors optimized a six-step pathway for producing n-butanol, an important solvent and drop-in biofuel, using a cell-free prototyping approach (iPROBE). iPROBE reduces the overall time to build pathways from weeks or months to a few days (around five in this case), providing the quick turnaround and large numbers of enzyme combinations that can enable successful use of machine learning. Several pathway variants were constructed in vitro and scored based on their measured butanol production through a TREE score which combines titer, rate, and enzyme expression. The challenge, however, lies in analyzing the sheer number of pathway combination possibilities. Testing only six homologs for the first four pathway steps at 3 different enzyme concentrations would result in 314,928 pathway combinations (strain genotypes). Even with the increased turnover provided by the cell-free approach, it would take years for typical analytical pipelines to exhaustively test the landscape of possible combinations. Therefore, a data-driven design-of-experiments approach was implemented using neural networks to predict optimized pathway designs (homolog sets and enzyme ratios) from an initial data set that could subsequently be tested. In this case the input for the neural network was the enzyme homologs used for each of the reaction steps and their corresponding concentrations. The response was the TREE score, and each instance was a pathway design.
The pathways predicted from the neural network model were able to improve butanol production scores over fourfold (~2.5 times higher titer, 58% increase in rate) compared to the base-case pathway. An initial data set of 120 instances (pathway designs) was used to train and test different neural network architectures consisting of 5–15 fully connected hidden layers and 5 to 15 nodes per layer. Genetic algorithms were used to suggest combinations of network architectures, and ten-fold cross validation was used to select the best. Once the model was built, the authors used a nonlinear optimization algorithm (Nelder-Mead simplex) to recommend pathway designs that optimized butanol production through the maximization of the TREE score. These machine learning recommendations resulted in 5 of the 6 top performing pathways, and outperformed 18 expert determined pathways selected based on prior knowledge, demonstrating the power of a data-driven design approach for cases in which design choices are numerous.
While the study by Karim et al. only reported 1 DBTL cycle, multiple cycles would have likely resulted in even better production pathways, and also provided more data instances for model training. Indeed, the neural network of 5–15 hidden layers developed by Karim et al. was relatively small compared to state-of-the-art deep neural networks, but this design was limited by having only 120 instances (pathway designs) to train on. If more data were to become available through more DBTL cycles, the neural network could have been made more complex by expanding its depth (hundreds of hidden layers), which would improve prediction performance (Fig. 4). This improved performance, however, comes at a cost: as the number of layers increases, the time to train the network (i.e. learning model weights and parameters) increases considerably. Moreover, the dense hidden layers of deep neural networks render them very difficult to interpret and infer possible mechanisms from. Hence a significant research thrust in machine learning involves new approaches to make models “explainable” (see Section 5.3) (Gunning, 2016; Gunning et al., 2019). The use of only 1–2 DBTL cycles seems to be the most common case in published projects (Denby et al., 2018; Alonso-Gutierrez et al., 2015; Opgenorth et al., 2019; Zhang et al., 2020). In our experience, this happens not because more DBTL cycles are not expected to be useful, but because results from a single DBTL cycle are often enough for a publication. Often, in the academic world, there is little incentive (or resources) to continue further.
2.3. Requirements for machine learning in metabolic engineering
Here we provide a practical guide on the immediate prerequisites to applying machine learning to metabolic engineering, in the next section we will discuss some practical considerations for experimental design once the machine learning project is in progress, and, in section 5.1, we discuss long term hurdles for the development of the discipline as a whole. In essence, four requirements need to be aligned for a successful application: data, algorithms, computing power and an interdisciplinary environment. Each of them is critical for a real impact.
Data needs to be abundant, non-sparse, high quality, and well organized. Training data needs to be abundant because machine learning algorithms depend critically on training data to be predictive. There is no prior biological knowledge embedded in them. In general, the more training data, the more accurate the algorithm predictions will be. Data augmentation (see section 2.2.1) can certainly help, and should be routinely used in metabolic engineering due to the scarcity of large data sets, but it is no substitute for experimental data. There is, however, no way to know a priori how much data will be enough. Different problems present different difficulty levels to being “learnt” (Radivojević et al., 2020), and this difficulty level can only be assessed empirically. A scaling plot of predictive accuracy vs. instances can be very helpful in this regard. Training data can be abundant but still sparse, depending on the phase space (Fig. 9) considered. A total of a hundred instances can be enough if only two input features are considered, or completely insufficient if a thousand input features are considered. The “curse of dimensionality” implies that the amount of data needed to support results in a statistically sound and reliable fashion often grows exponentially with the dimensionality (Fig. 4). The data must be high-quality in the sense that it must avoid biases due to inconsistent protocols and provide quantification for repeatability (see section 2.4). Both goals can be systematically achieved through automation (see section 5.2). Data needs to be well organized, following standards and ontologies, and must include the corresponding metadata (see section 2.4). The alternative is that data analysts will spend 50–80% of their effort organizing the data and metadata for analysis, mining their efforts (Lohr, 2014). Since data analysts might be the most effective effort multiplier in your team (Nielsen and Keasling, 2016), and possibly the most expensive (Metz, 2018), it is very useful to optimize their effort.
 Fig. 8
Fig. 8 Fig. 9
Fig. 9While there are many machine learning algorithms to choose from (Fig. 10), there is no clear best algorithm for every situation. Indeed there is a famous theorem (the no free lunch theorem, NFLT) that proves (under some conditions) that no single algorithm is most effective for every type of problem (Wolpert, 1996). While the utility of the NFLT for machine learning has been cast in doubt (Giraud-Carrier and Provost, 2005 ), the standard approach remains to try as many algorithms as possible and compare their results. In this effort, it is very useful to count on libraries that collect a large variety of algorithms and have standardized input, output and other standard procedures (e.g. cross-validation). The most popular among them is, without a doubt, scikit-learn (Pedregosa et al., 2011), a python library that comprises a very wide selection of machine learning methods, is well documented, and easy to use (Fig. 10). These features combined with its open source nature, and its compatibility with Jupyter notebooks (Kluyver, 2016), which facilitate reproducibility and communication, make it our top recommendation for beginners. Furthermore, the open source nature and wide use of scikit-learn means that there are several tools that leverage it to combine and test methods. Tree-based pipeline optimization tool (TPOT), for example, automatically combines all the available algorithms and preprocessing steps in scikit-learn to choose the best option (Olson et al., 2016). Another example is the Automated Recommendation Tool (ART), which leverages scikit-learn, ensemble modeling, and bayesian inference to provide uncertainty quantification for predictions (Radivojević et al., 2020). A proprietary alternative is to use Matlab, for which a machine learning toolbox is available (Ciaburro, 2017), with possible educational discounts. For artificial neural networks, the best supported (and free) frameworks are TensorFlow and Pytorch, backed by Google and Facebook respectively. Keras, a framework focused on providing a simple interface for neural networks, is now the official high-level front-end for TensorFlow (Géron, 2019). Keras has its own hyperparameter tuner, Keras Tuner (O'Malley et al., 2019), and an extremely simple interface for DL with Keras and TensorFlow, AutoKeras (Jin et al., 2019).
 Fig. 10
Fig. 10Computation is another key element, particularly for large amounts of data. Whereas the libraries above (Scikit-learn, Matlab toolbox, Tensorflow, Pytorch) can be run on a standard laptop (e.g. 2018 Macbook Pro, 3.5 Ghz Intel Core i7, 16 GB RAM), as more training data is added this may be insufficient. This is particularly the case for deep neural networks using Tensorflow or Pytorch, which will benefit from the parallelization obtained through Graphics Processing Units (GPUs). The need to scale up all these Python frameworks for high performance computing (HPC) or deployment on cloud computing environments (e.g. Amazon EC2, Microsoft Azure, and Google's Cloud Platform) has promoted the development of several parallel and distributed computing backends for data analysis and machine learning, such as Ray, Spark, and Dask (Rocklin, 2015). Furthermore, as the general applicability of AI has become more evident, new processor architectures are being created specifically for neural network machine learning, including Google's Tensor Processing Unit (TPU), Nvidia's V100 and A100, Graphcore's Intelligence Processing Unit (IPU), and a variety of FPGA-based solutions.
Since very few people master both machine learning and metabolic engineering, interdisciplinary collaborations are truly necessary. Machine learning practitioners and metabolic engineers are trained very differently, however, and this can produce significant friction (see section 5.1). Both disciplines profess different cultures, which are reflected in how they solve problems, but also which problems are prioritized. It is, hence, very important to foster an inclusive work environment that integrates and values contributors with very different skills, and does not penalize knowledge gaps. It is also important to be very clear about the interfaces: which exchanges (e.g., data, designs, predictions) are expected, and when, in order for both sides to be effective.
2.4. Practical considerations for implementing machine learning
As in the case of a genetic selection or screen, machine learning requires careful experimental planning to make it effective. An experimental design that ignores its basic assumptions (e.g., instances are independent and identically distributed) will result in a random walk over possible designs with the same (or even worse) results as a trial-and-error approach.
Here, we offer a succinct list of recommendations to consider when planning to use machine learning to guide bioengineering:
-
●
Choose the right objective/response. When a response for the algorithm is chosen, you are entering a Faustian bargain with your algorithm: it will try to optimize it to the detriment of everything else (Riley 2019) . For example, setting final titer as the response might provide high titers in the end for a production strain, but at rates so slow that the result is of little practical use. In the case of Karim et al., (see section 2.2.2), the response was a carefully selected mixture of titer, rate, and enzyme expression precisely for this reason. Deciding on the right response is a bit of an art, and less trivial than often assumed. Be careful what you ask the algorithm for, because you may get it!
-
●
Choose inputs that truly predict your response. Performing small, directed experiments in the lab to verify that the response of interest (e.g. a phenotype) is affected by a given input (e.g. a treatment) can save a significant amount of time and headaches later in the DBTL cycle, by limiting the number of inputs (and the overall complexity of the model) to terms that matter. Omitting this step might give rise to a frustrating chase of a red herring in the form of statistical noise, or cause serious challenges to the interpretability of the model.
-
●
Choose actionable inputs that can be measured. The machine learning process will require you to change your inputs in order to achieve the desired goal (e.g. increase production). Hence, these inputs need to be experiment variables that can be easily manipulated. Since you will need to assess whether you indeed reached the recommended targets, it is highly desirable that these inputs can be easily measured. For example, it is generally better to use as inputs promoter (Zhang et al., 2020) or enzyme choices (Karim et al., 2020), rather than protein levels (Opgenorth et al., 2019). Promoter or enzyme choices are entirely under the metabolic engineer's control, and their effects on expression may be verified via sequencing; whereas certain target protein levels may be difficult to reach, and usually require specialized mass spectrometry methods to verify.
-
●
Choose very carefully how many experiment variables you would like to explore. Choosing too many variables (i.e. input features, Fig. 3) can make the corresponding phase space too large for machine learning to explore in a reasonable amount of DBTL cycles. Choosing too few variables might mean missing important system configurations (e.g. if protein X is not chosen and it needs to be downregulated to improve production, it will be impossible to find the optimum). As a very crude rule of thumb, you should budget for around at least 100 instances per 5–10 variables. This, of course, depends on the difficulty presented by the problem being learnt: more difficult problems will need more instances per variable, whereas easier problems will require less instances per variable.
-
●
Verify that your experiment variables can be independently acted upon. Whole-operonic effects can make this unexpectedly difficult (Opgenorth et al., 2019). For example, if recommendations require protein A concentration to be increased three-fold and protein B to be decreased by a factor of two to improve production, but a strong promoter for protein A also produces an increase in protein B, it will be difficult to reach the target protein profile. Hence, modular pathway designs (Boock et al., 2015) that ensure that the full input phase space can be fully explored are highly recommended. Systematic part characterization involving large promoter libraries with a variety of tested relative strengths are a fundamental tool in this endeavor.
-
●
Design your experiment to start with ~100 instances for the initial DBTL cycle. Although there are examples of success stories with less than a hundred instances as starting points (Radivojević et al., 2020), this outcome cannot be guaranteed. Actual success depends on the complexity of the problem (Radivojević et al., 2020), and this complexity can only be gauged by testing predictive accuracy as data sets increase. By starting with ~100 instances, one ensures some progress even if predictions are not accurate: this amount of instances goes a long way to ensure statistical convergence. The alternative is a non-predictive model and little understanding whether the problem is lack of data (instances), or other design problems (Opgenorth et al., 2019). Consider automating as much of your process as possible so as to guarantee enough instances. This automation may seem an unnecessary hassle, but it will pay off in the long run.
-
●
Sample the initial phase space as widely as possible. Ensure that you cover wide ranges for both input and response variables. Strive to include both bad (e.g. low production) and intermediate results as well as good ones (e.g. high production), since this is the only way that the algorithms can learn to distinguish the inputs needed to reach any of these regimes. The Latin Hypercube (McKay et al., 1979) is a good choice to choose starting points, but other options are also available.
-
●
Consider uncertainty, as well as predicted response, when choosing next steps. As the need to quantify prediction uncertainty becomes more recognized in the biological sciences (Begoli et al., 2019), more algorithms provide it along with response predictions (Radivojević et al., 2020). Using this information can improve the whole process. Choose some recommendations with the lowest possible uncertainty even if the predicted outcome is not so desirable (e.g. low production), so as to establish trust in the approach (see sociological hurdles in section 5.1). Choose some recommendations with large uncertainty even if the predicted outcome is not desirable so as not to miss unexpected opportunities. In addition, to obtain an empirical view of how uncertainty in the data affects the accuracy of predictions, it may be instructive to create simulated, in silico “ground truth” data sets displaying different levels of noise in order to test the performance of the machine learning algorithm.
-
●
Avoid biases created through inconsistent protocols and beware of hidden variables. Machine learning algorithms learn to map an input to a response (Fig. 3). If different DBTL cycles produce different results for reasons that are not reflected in the input (hidden variables (Riley 2019)), the algorithms will provide poor predictions. Such uncontrolled variables can easily arise in biological data due to lab temperature or climate fluctuations, reagent batch differences, undetected culture mutations, “edge effects” in plate-based assays, and equipment drift. These effects should be assessed and eliminated as part of the experimental design, and is one of the key topics of communication for bench and computational scientists to empower downstream data analysis and predictions. Machine learning can also help by performing simple checks: if an algorithm can predict which well or batch sample the data came from, that means they unduly influence the response. Lack of repeatability is the main stumbling block of machine learning.
-
●
Add experimental controls to test for repeatability. Since ensuring repeatability is among the top requirements for machine learning to be successful, it is important to test and quantify it often. Batch, instrument, and operator effects are often the first principal component of data. These effects can be detected by including a few controls of known response in every experiment (e.g., 2–3 base strains in every DBTL cycle). While this approach consumes valuable analytical resources, it ensures that the data can be trusted and does not need to be discarded, saving substantial labor during modeling and analysis.
-
●
Plan for several DBTL cycles. Machine learning algorithms shine when they can dynamically probe your system, since they are designed to learn from data interactively. While results can be obtained using two DBTL cycles, they are not comparable to what >5 cycles can provide (Radivojević et al., 2020). If only a limited budget of, e.g. 100 instances, is available, it is better to start with a strong first cycle and several weaker ones (e.g. 40 instances for cycle 1, then six 10 instance cycles) than the usual two DBTL cycle study (e.g. 60 instances for first cycle, 40 instances for the second one).
-
●
Standardize your data and metadata. Taking machine learning for metabolic engineering seriously requires large amounts of high quality data. Hence, it is advisable to store it in a standardized manner. There are a variety of data repositories available for this purpose: e.g., the Experiment Data Depot (Morrell et al., 2017), the Inventory of Composable Elements (ICE) (Ham et al., 2012), DICOM-SB (Sainz de Murieta et al., 2016), SynBioHub (McLaughlin et al., 2018), ProteomeXchange (Vizcaíno et al., 2014), MetaboLights (Haug et al., 2013), BioGraph-IIn (Gonzalez-Beltran et al., 2013), the Nature Scientific Data journal (“Open for business,” 2017), to name a few. Moreover, a labeled data set of high quality is a significant resource for the community, and is more likely to be cited.
-
●
Be careful about how you split your data for cross-validation. Cross-validation of your model (Fig. 3), assumes data sets are independent and identically distributed (iid). This assumption is basic for machine learning, and presumes that both validation and training sets stem from the same generative processes and have no memory of past generated samples. However, it can be violated in practice due to temporal effects on biological systems or group effects during sample processing (Riley 2019). In these cases, alternatives to random splitting need to be considered. Sheridan (2013), for example, showed that randomly splitting compound libraries used for drug discovery overestimated their model's ability to successfully predict drug candidates. The reason for this difference is that compounds added to the public record at particular dates shared higher structural similarity, resulting in models that had already “seen” compounds in the test set when randomly split. Similar considerations need to be made when sample generation occurs in a biased manner, which is quite common in biological experiments. For example, “batch effects” can be avoided by splitting the data first by group (e.g. each batch) to ensure the same group is not represented in both testing and training sets (see scikit-learn group k-fold). Do only worry about this effect if you have a large data set (>100 instances).
Perhaps the best way to get familiar with machine learning, and its potential and limitations, is to experiment with it in a tutorial. The recently published Automated Recommendation Tool (Radivojević et al., 2020) includes three synthetic data sets, three real data sets and a software package that can be used for this purpose. Furthermore, some of these cases are explained in detail in several Jupyter notebooks contained in the github repository (https://github.com/JBEI/ART/tree/master/notebooks), and can be used as tutorials.
3. Applications of machine learning to metabolic engineering
Although application of machine learning in metabolic engineering is nascent, early studies have already shown its potential use for accelerating bioengineering. Here, we highlight examples where machine learning is being used to improve different stages of the metabolic engineering development cycle: gene annotation and pathway design, pathway optimization, pathway building, performance testing, and production scale-up (Table 1). We focus on prime examples that best epitomize the potential of machine learning in metabolic engineering, rather than an exhaustive list of applications. The reason for this decision is that this list is quickly growing and might be outdated soon, and there are recent reviews on the topic that provide that information (Kim et al., 2019; Presnell and Alper, 2019; Volk et al., 2020). We also discuss key challenges and opportunities when applying machine learning for metabolic engineering, with particular focus on practices that could formalize data-driven approaches.
Table 1. Machine learning applications for metabolic engineering.
| Task | Application | Input features | Algorithm | Response | Ref. |
|---|---|---|---|---|---|
| Identify ORF | Identify signal peptide (SignalP 5.0) | 20,758 protein amino acid sequence | deep RNN | presence or absence of signal peptide | Armenteros et al. (2019) |
| ORF prediction (DeepRibo) | 626,708 candidate ORF DNA sequences and ribo-seq signal (alignment file) from 7 species | RNN and CNN | translation initiation site and translated open reading frames | Clauwaert et al. (2019) | |
| ORF prediction (REPARATION) | 67,158 candidate ORF DNA sequences and ribo-seq signal (alignment file) from 4 species | random forest | translation initiation site and translated open reading frames | Ndah et al. (2017) | |
| Annotate ORF | Annotate enzyme (DeepEC) | 1,388,606 protein sequences and 4669 EC numbers | CNN | enzyme commission (EC) numbers | Ryu et al. (2019) |
| Annotate enzyme (ECPred) | 11,018 protein amino acid sequences, subsequence extraction and peptide physicochemical properties | ensemble classifier (BLAST-kNN, Pepstats-SVM and SPMap) | EC numbers | Dalkiran et al. (2018) | |
| Annotate enzyme (DEEPre) | 22,168 protein amino acid sequence | CNN and RNN | EC numbers | Le et al., 2018 | |
| Annotate enzyme (EnzyNet) | 63,558 protein sequences represented as voxel-based protein spatial structure | CNN | EC numbers | Amidi et al., 2018 | |
| Enzyme & Pathway design | Automated enzyme search | 10,951 compounds and 6556 reactions. Features represented as reaction signature and enzyme amino acid sequence | support vector machines | positive or negative enzyme-reaction pairs | Faulon et al. (2008) |
| Automated enzyme search | 7318 reactions and 9001 enzymes. Features represented as reaction signature and enzyme amino acid sequence pair | gaussian process model | positive or negative enzyme-reaction pairs, and | Mellor et al. (2016) | |
| Michaelis constant KM (substrate affinity) | |||||
| Directed evolution | 805 protein amino acid sequence variants | linear, kernel, neural network, and ensemble methods | protein fitness | Wu et al. (2019) | |
| Directed evolution | 585,199 protein amino acid sequence variants | partial least-squares linear regression | bacterial halohydrin dehalogenase productivity | Fox et al. (2007) | |
| and multivariate optimization | |||||
| Directed evolution | 218 protein amino acid sequence variants | gaussian process | fluorescent protein color | Saito et al. (2018) | |
| Directed evolution | 4716 protein amino acid sequence variants | gaussian process | protein thermostability | Romero et al. (2013) | |
| Rational protein design (UniRep) | ∼24 millon protein amino acid sequence | RNN | protein feature representation | Alley et al. (2019) | |
| Rational protein design | 96 protein amino acid sequence variants (UniRep encoding) | UniRep pretraining + linear regression (ridge, lasso-lars, ensemble) | protein fitness | Biswas et al. (2020) | |
| Rational protein design (BioSeqVAE) | protein amino acid sequence | residual neural networks | protein representation | Costello and Garcia Martin, 2019 | |
| Synthetic pathway design (RetroPath RL) | N/A | Monte Carlo Tree Search reinforcement learning | metabolic pathway | Koch et al. (2020) | |
| Pathway optimization | Promoter design | 675,000 constitutive and 327,000 inducible promoter sequences | CNN | gene expression activity | Kotopka and Smolke, 2020 |
| Promoter design | 100 mutated promoter and RBS sequences | neural network | promoter strength | Meng et al. (2013) | |
| Promoter design | promoter amino acid sequence | neural network | promoter strength | Tunney et al., 2018 | |
| Riboswitch design | biophysical properties (entropy, stem melting temperature, GC content, length, free energy, etc) from 96 riboswitch aptamer sequences | Random forest and CNN | dynamic range of gene expression between ON/OFF states | Groher et al. (2019) | |
| Plasmid design (SelProm) | 120 plasmid sequences | partial least-squares regression | promoter strength, induction time, inducer concentration | Jervis et al. (2019a) | |
| multi-gene pathway optimization (MiYA) | 24 strains, different promoter combinations | neural network | β-carotene and violacein production | Zhou et al. (2018) | |
| multi-gene pathway optimization | 156 strains, different RBS sequences | support vector machines and neural network | limonene production titer | Jervis et al. (2019b) | |
| multi-gene pathway optimization(BioAutomata) | 136 strains, different promoter and RBS sequences | gaussian process and Bayesian optimization | lycopene production titer | HamediRad et al. (2019) | |
| (Automated Recommendation Tool (ART) | promoter combinations, multi-omics data, etc | Bayesian ensemble model | chemical production titer, rate, yield | Radivojević et al. (2020) | |
| multi-gene pathway optimization | 250 strains, different promoter combinations | probabilistic ensemble model (ART) and Bayesian optimization (EVOLVE algorithm) | tryptophan production titer | Zhang et al. (2020) | |
| Multi-gene Pathway optimization | 12 strains, 4 biological replicates per strain | Ensemble model (random forest, polynomial, multilayer perceptron, TPOT meta-learner) | Dodecanol production | Opgenorth et al. (2019) | |
| CRISPR | sgRNA Scorer 2.0, CRISPR activity | 430 sgRNA sequences | support vector machine | sgRNA on-target activity | Chari et al. (2017) |
| Azimuth, CRISPR activity | 4390 sgRNA sequences | support vector machine with logistical regression | sgRNA on-target and off-target activity | Doench et al. (2016) | |
| Seq-DeepCpf1, CRISPR activity | 16,292 sgRNA sequences | CNN | sgRNA on-target activity | Kim et al. (2018) | |
| Elevation, CRISPR activity | 299,387 sgRNA–target pairs | gradient boosted regression trees | sgRNA off-target activity | Listgarten et al. (2018) | |
| CRISPR activity | 294,534 sgRNA-target pairs | CNN and deep feedforward neural network | sgRNA off-target activity | Lin and Wong (2018) | |
| DeepCRISPR, CRISPR activity | 0.68 billion sgRNA sequences (unlabeled pre-training data) ∼160,000 sgRNA-target pairs (off-target data sets) ∼200,000 sgRNA seq uences (on-target data set) | deep convolutional denoising neural network and CNN | sgRNA on-target and off-target activity | Chuai et al. (2018) | |
| Outlier Detection | Novelty and Outlier Detection | Any training data set | isolation forest, local outlier factor, one-class SVM, and elliptic envelope | outlier identification | https://scikit-learn.org/stable/modules/outlier_detection.html |
| Omics Data Processing | Prosit, peptide identification | 550,000 tryptic peptides | bi-directional RNN | peptide chromatographic retention time and tandem mass spectra | Gessulat et al. (2019) |
| Peakonly, metabolite peak detection | 4000 regions of interest, labeled as noise, one or more peaks, or uncertain peak | CNN | peak detection + integration (peak area) | Melnikov et al. (2020) | |
| Bioprocess Control & optimization | Bioprocess optimization | 69 fed-batch fermentations, 13 process features (fermentation conditions, inoculum conditions, media variables) | three-step optimization method using decision trees, neural network, and hybrid genetic algorithm | maximum cell concentration, product concentration, and productivity | Coleman et al. (2003) |
| Bioprocess optimization | 25 fed-batch fermentations, 11 process features (temperature, induction strength, growth rate, process variables) + spectroscopic information | data preprocessing, random forest and neural network | cell dry mass, recombinant soluble protein conc., inclusion bodies conc. | Melcher (2015) | |
| Bioprocess optimization | 27 batch fermentations, 7 process features (time, pH, temperature, kLa, biomass, xylose, glycerol) | regression and neural network coupled to genetic algorithm for optimization | xylitol production | Pappu and Gummadi (2017) | |
| Bioprocess control & real-time optimization | continuous bioreactor, 24 h duration with measurement/action every 5 min | Neural network fitted Q-learning algorithm (reinforcement learning) | control species biomass ratio; maximize product yield | Treloar et al. (2020) | |
| Process control | 600 temperature measurement/action timesteps (episodes) | Model Predictive Control (MPC) guided deep deterministic policy gradient (reinforcement learning). Policy parameterized by neural network | Reactor (CSTR) temperature control | Xie et al., 2020 | |
| Real-time process optimization | 500 measurement/action episodes | Policy gradient parameterized by a recurrent neural network. Transfer learning from offline training on mechanistic model | Maximize product yield (phycocyanin) | Petsagkourakis et al., 2020 | |
| Process control | 21 measurement/action episodes | multi-step action Q-learning controller based on fuzzy k selector | ethanol concentration control | Li et al., 2011 |
Notes: RNN = recurrent neural network; CNN = convolutional neural network.
3.1. Machine learning for design
The goal of metabolic engineering design is to develop DNA parts and assembly instructions to synthesize metabolic pathways and produce a desired molecule (Nielsen and Keasling, 2016; Woolston et al., 2013). This requires completion of several tasks, including gene annotation, pathway reconstruction and design, as well as metabolic flux optimization, which currently rely heavily on domain expertise and enjoy little standardization (Nielsen and Keasling, 2016). Application of machine learning can improve the accuracy and speed of these tasks, offering a standardized approach that fully leverages experimental data.
3.1.1. Pathway reconstruction and design
Locating and annotating protein encoding genes in a genome sequence is essential for metabolic pathway reconstruction and design. This is conventionally done bioinformatically, for example using Hidden Markov Models (HMMs) (Finn et al., 2011; Kelley et al., 2012; Yoon, 2009). Initially, genes are identified in a genome by searching for known protein coding signatures (e.g. Shine-Dalgarno sequences), and this is followed by annotation based on sequence homology searches against a database of previously characterized proteins. More recently, however, deep learning approaches have been used to identify and functionally annotate protein sequences in genomes by leveraging large high-quality experimental data sets (Armenteros et al., 2019; Clauwaert et al., 2019; Ryu et al., 2019). DeepRibo, for example, uses high-throughput ribosome profiling coverage signals and candidate open reading frame sequences (input features) to train deep neural networks to delineate expressed open reading frames (response is part of predicted ORF or not for every nucleotide) (Clauwaert et al., 2019). This approach showed more robust performance compared to a similar tool, REPARATION (Ndah et al., 2017), that uses a random forest classifier instead of deep neural networks. DeepRibo also improved prediction of protein coding sequences in different bacteria (e.g. Escherichia coli and Streptomyces coelicolor) compared to RefSeq annotations, including higher identification of novel small open reading frames commonly missed by sequence alignment algorithms. Another example is DeepEC, which takes a protein sequence as input and predicts enzyme commission (EC) numbers as output with high precision and throughput using deep neural networks (Ryu et al., 2019). A data set containing 1,388,606 expert curated reference protein sequences and 4669 enzyme commission numbers (Swiss-Prot (Bairoch and Apweiler, 2000) and TrEMBL (UniProt Consortium, 2015) data sets) was used to train the deep neural networks, which improved EC number prediction accuracy and speed compared to 5 alternative EC number predictions tools, including CatFam (Yu et al., 2009), DETECT v2 (Nursimulu et al., 2018), ECPred (Dalkiran et al., 2018), EFICAz2.5 (Kumar and Skolnick, 2012), and PRIAM (Claudel-Renard et al., 2003). DeepEC was also shown to be more sensitive in predicting the effects of protein sequence domain and binding site mutations compared to these tools, which could improve the accuracy of annotating homologous proteins that have mutations with previously unknown effects on function (e.g. from metagenomic data sets).
The design of metabolic pathways involves identifying a series of chemical reactions that produce a desired product from a starting substrate, and selecting different enzymes that catalyze each reaction. While nature has evolved many pathways for producing diverse molecules, the known and characterized biochemical pathways can still be insufficient to produce certain molecules of interest, especially non-natural compounds or secondary metabolites. Therefore, retrosynthesis methods that start with a desired chemical and suggest a set of chemical reactions that could produce it from cellular metabolite precursors are being pursued to design new metabolic pathways (Lin et al., 2019; Lee et al., 2019). The latest and most sophisticated of these methods use generalized reaction rules to describe possible biochemical transformations (Delépine et al., 2018; Kumar et al., 2018). However, the number of possible reaction combinations is intractable since it grows combinatorially with the number of reactions. Choosing the right reaction combination is a non-trivial problem, which is typically tackled via optimization or heuristic methods. A possible solution to this search problem comes from solving the same problem in organic synthesis, through the use of deep neural networks (Segler et al., 2018). Segler et al. preprocessed 12.4 million reaction rules from the Reaxys chemistry database to train three deep neural networks implemented within a Monte Carlo tree search (heuristic search algorithm used in decision making) to discover retrosynthesis routes for small molecules. This deep learning approach found pathways for twice as many molecules, thirty times faster than traditional computer-aided searches (Segler et al., 2018). The predicted synthesis routes better adhered to known chemical principles than traditional computer-aided searches and could not be differentiated by expert organic chemists compared to synthesis routes taken from the literature, highlighting the potential of deep learning to be applied for metabolic retrosynthesis (or retrobiosynthesis). Indeed, a similar Monte Carlo Tree Search method has recently been extended to predict synthetic pathways within biological systems (RetroPath RL), enabling systematic pathways design for metabolic engineering (Koch et al., 2020).
Pathways designed via retrosynthesis still face the difficult challenge of finding enzymes for novel biochemical reactions, for which no enzyme is known. In this case, the solution involves enzymes that may catalyze the novel reaction through enzyme promiscuity, or new enzyme functions must be designed or evolved that perform the desired chemistry. While chemoinformatic techniques (e.g. density functional theory, DFT, and partitioned quantum mechanics and molecular mechanics, QM/QM) can be used to predict the interaction between metabolites and proteins in silico (Alderson et al., 2012), these techniques are computationally intensive and require substantial domain expertise. Therefore, the task of searching for promiscuous enzymes is increasingly being performed using more general and computationally efficient techniques from machine learning. For example, given a reaction and enzyme pair instance, Support Vector Machines (Faulon et al., 2008) and Gaussian Processes (Mellor et al., 2016) have been developed to predict whether the enzyme catalyzes the reaction, with the latter model having the added benefit of providing uncertainty quantification. These models predict positive or negative enzyme reaction pairs from protein sequences (e.g. K-mers) and reaction signatures (e.g. functional groups, chemical transformation properties) (Carbonell and Faulon, 2010) by learning patterns about promiscuous enzyme activities through training. They can also be applied to predict substrate affinity for proteins (Km values) (Mellor et al., 2016), an important kinetic parameter for determining enzyme activity, which is difficult and time consuming to measure experimentally. This is critical for pathway design as sequences with the most desirable kinetic properties can be selected when multiple candidates catalyzing a given reaction are available.
In the case that no enzyme can be found for a target reaction, new enzymes may be designed or discovered through protein engineering. A common laboratory method for protein engineering is directed evolution, where beneficial mutations accumulate in a protein through iterative experimental rounds of mutation and selection until the desired protein function is achieved (Yang et al., 2019). In essence, a series of local searches (via sequence mutation and screening) are performed on an enormous and highly complex functional landscape with the hope of finding a local optima (i.e. protein variant with desired properties). However, experimental approaches can only explore an infinitesimal part of this landscape and computational approaches are needed to guide experimental efforts. Machine learning can be used to guide directed evolution and decrease the number of experimental iterations needed to obtain a protein with the desired function. This is achieved by leveraging previous screening data to learn a protein's sequence-function landscape and predict new sequence libraries that contain variants with higher fitness. For example, instead of experimentally performing sequential single point mutations or recombining mutations found in best variants (common directed evolution approaches), Wu et al. (2019) trained a machine learning model to perform in silico evolution rounds that ranked new protein variants by predicted fitness for experimental testing. Instead of relying on a single machine learning method, multiple models (linear, kernel, neural network, and ensemble) were trained in parallel, and the ones showing the highest accuracy were used to perform in silico evolution rounds (Wu et al., 2019). This enabled deeper exploration of the possible variant functional landscape, resulting in the successful evolution of an immunoglobulin-binding protein and a putative nitric oxide dioxygenase from Rhodothermus marinus. ML-assisted directed evolution has also been used to maximize enzyme productivity (Fox et al., 2007), change the color of fluorescent proteins (Saito et al., 2018), and optimize protein thermostability (Romero et al., 2013) making it a promising approach for searching large sequence-function spaces in an efficient manner for proteins variants with desired properties.
In addition to directed evolution, deep learning has also recently been applied for the rational design of proteins (Alley et al., 2019; Biswas et al., 2020; Costello and Garcia Martin, 2019). For example, Alley et al. (2019) developed UniRep, which uses recurrent neural networks to learn an internal statistical representation of proteins that contained physicochemical, organism, secondary structure, evolutionary and functional information, by training on 24 million UniRef50 (Suzek et al., 2015) amino acid sequences (instances). The resulting representation was applied to train models (random forest or sparse linear model) using UniRep encoded proteins that predicted the stability of a large collection of de novo designed proteins and also the functional consequence of single point mutations on wild-type proteins. UniRep encoding was also used to optimize the function of two fundamentally different proteins (to wild-type), a eukaryotic green fluorescent protein from Aequorea victoria, and a prokaryotic β-lactam hydrolyzing enzyme from Escherichia coli, highlighting the generalizability of this approach for rational protein engineering (Biswas et al., 2020). Other generative models based on deep learning have been used to suggest protein sequences with desired functionality and location (Costello and Garcia Martin, 2019).
3.1.2. Pathway optimization
Following pathway design, metabolic flux optimization is required to maximize product titers, rates, and yields (TRY). In this endeavor, machine learning provides an orthogonal approach to computational approaches leveraging flux analysis and genome-scale models, which have been successfully used in the past to increase TRY (Maia et al., 2016). The combination of both approaches has the potential to be more effective than each of them separately (see section 4 for a discussion).
A common approach to increase TRY involves fine tuning gene expression through the modification of promoter and ribosome binding site (RBS) sequences. Despite decades of progress in understanding the regulatory mechanisms controlling gene expression (Snyder et al., 2014), quantitative prediction of gene expression based on sequence information remains challenging. While computational models do exist to predict gene expression (Leveau and Lindow, 2001; Salis et al., 2009; Rhodius and Mutalik, 2010), they rely on a comprehensive understanding of transcription and translation processes. This knowledge is often unavailable, especially for non-model organisms. Therefore, many gene expression optimization efforts rely on trial-and-error experimental approaches based on promoter and RBS library screening (Choi et al., 2019), that also suffer from the large combinatorial space of possible sequences.
Machine learning has also guided the design of promoter and RBS sequences in a data-driven manner for improved control of gene expression. In particular, neural networks have been used to predict gene expression output from input promoter sequences or coding regions (Kotopka and Smolke, 2020; Meng et al., 2013; Tunney et al., 2018). Meng et al. (2013) used a simple neural network trained with 100 mutated promoter and RBS sequences as inputs to predict promoter strength (response). This machine learning model outperformed mechanistic models based on position weight matrix or thermodynamics methods (Leveau and Lindow, 2001; Salis et al., 2009; Rhodius and Mutalik, 2010), and was able to optimize heterologous expression of a small peptide BmK1 (used in traditional Chinese medicine) and the dxs gene involved in the isoprenoid production pathway (Meng et al., 2013). Additionally, optimization of promoter strength and inducer concentration/time has been achieved using partial least squares regression (Jervis et al., 2019a), whereas prediction of riboswitch dynamic range from aptamer sequence biophysical properties has been achieved using a combination of random forests and neural networks (Groher et al., 2019). In this latter riboswitch design example, instead of directly using sequence information to train the random forest, the authors calculated known riboswitch biophysical properties from aptamer sequences (entropy, stem melting temperature, GC content, length, free energy, etc.) and used these as input features for model training, in order to predict switching behavior. This allowed for the interpretation of which input features were most important to the model prediction using variable importance (e.g. melting temperature was more important than free energy), enabling inferences on possible mechanisms.
More recently, machine learning models have been used to optimize multi-step pathways for chemical production (Zhou et al., 2020). For example, Zhou et al. (2018) used neural network ensembles to improve a 5-step pathway for violacein production (pharmaceutical) by selecting promoter combinations to tune gene expression. Using an initial training set of only 24 strains (out of a possible 500) containing different promoters for each gene, the model predicted a new strain that improved violacein titer by 2.42-fold after only 1 DBTL iteration. Their ensemble approach allowed top producing strains to be predicted from a combination of over 1000 ANN, which improved model accuracy and also allowed optimization of violacein based on both titer and purity. In another example, Opgenorth et al., (2019) used an ensemble of four different models (random forest, polynomial, multilayer perceptron, TPOT meta-learner) to optimize a 3-step pathway for dodecanol production from two DBTL cycles. The model was trained using data generated from 12 strains (48 data points total) with different RBS sequences for each gene, where an optimization step was used to recommend improved strain designs to build and test in the second cycle. Additional machine learning models have guided the optimization of multi-gene pathways, including limonene production in E. coli using support vector regression (Jervis et al., 2019b), lycopene synthesis in E. coli using gaussian processes (HamediRad et al., 2019), and tryptophan production in S. cerevisiae using ensemble models (Zhang et al., 2020). Together, these examples highlight the potential of systemically leveraging high-throughput strain construction, testing, and machine learning to optimize multi-step pathway expression for improving product TRY.
To enable broader use of ML-driven pathway optimization and design by the metabolic engineering community, Radivojevic et al. (Radivojević et al., 2020) developed the Automated Recommendation Tool (ART). ART is specifically tailored to the needs of the metabolic engineering field: effective methods for small training data sets and uncertainty quantification. ART's ability to quantify uncertainty enables a principled way to explore areas of the phase space that are least known, and is of critical importance to gauge the reliability of the recommendations. We expect that further development of tools tailored to the specific needs of the field will enable broader application of machine learning.
3.2. Machine learning for building and testing cellular factories
Machine learning can also be used to improve the tools that build and test cellular factories. A major challenge in gene editing using CRISPR-Cas technologies, for example, is predicting the on-target knockout efficacy and off-target profile of single-guide RNA (sgRNA) designs. Several approaches exist to make these predictions, including alignment-based methods (Aach et al., 2014), hypothesis-driven methods (Heigwer et al., 2014; Hsu et al., 2013), and classic machine learning algorithms (i.e. non-deep learning) (Chari et al., 2017; Doench et al., 2016). However, their generalizability has been limited by the small size and low quality (high noise) of the training data. Higher-throughput screening methods combined with deep learning have recently improved the accuracy and generalizability of sgRNA activity prediction tools. For example Kim et al (Kim et al., 2018), developed DeepCpf1, which predicts on-target knockout efficacy (indel frequencies) using deep neural networks trained on large-scale sgRNA (AsCpf1) activity data sets. While previous machine learning tools had been trained on medium-scale data (1251 target sequences), the authors high-throughput experimental approach generated a data set of indel frequencies for over 15,000 target sequence compositions, which was sufficient to train deep neural networks. Seq-DeepCpf1 was shown to outperform conventional ML-based algorithms, and steadily increased in performance as training data size increased, highlighting the value of data sets with >10,000 high-quality training instances. Seq-DeepCpf1 was also extended by considering input features other than target sequence composition known to affect sgRNA activity (in this example, chromatin accessibility (Jensen et al., 2017)) that further improved prediction accuracy and performance on independently collected data sets from other cell types (a metric of model generalizability). This highlights the value of expanding input features beyond the obvious choice.
In addition to predicting on-target knockout efficacy, the off-target profile of sgRNA activity is also important to forecast, in order to prevent undesirable perturbations that result in genomic instability or functional disruption of otherwise normal genes. This has been performed using both regressive models and deep neural networks (Listgarten et al., 2018,Lin and Wong, 2018). To combine on-target knockout efficacy and off-target profile predictions into one tool Chuai et al (Chuai et al., 2018), developed DeepCRISPR. DeepCRISPR uses both an unsupervised deep representation learning technique and deep neural networks to maximize on-target efficacy (high sensitivity), while minimizing off-target effects (high specificity). Unsupervised representation learning allows DeepCRISPR to automatically discover the best representation of input features from billions of genome-wide unlabeled sgRNA sequences, instead of specifying what input features should look like (e.g. target sequence composition). This sgRNA representation was then used when training a deep neural network using labeled data consisting of target sequences and epigenetic information (input features) to predict both on-target and off-target activities (responses). Overall, DeepCRISPR outperformed classic machine learning methods and exhibited high generalizability to other cell types, highlighting the value of unsupervised representation learning to automate feature identification.