1. Introduction
Since the discovery and development of the first vaccine and the first antibiotic in 1796 and 1928, respectively, mortality rates due to infectious diseases, such as smallpox or cholera, have markedly declined. Life expectancy has dramatically increased from an average of 47 to 80 years in the developed world [1], [2], [3]. Consequently, cancer has risen in priority as one of the most catastrophic illnesses plaguing the world. In the United States—a country with some of the most advanced cancer-treatment therapies—these diseases are still the second leading cause of death; they claimed 595 930 lives in 2015, which is just 37 912 fewer people than those killed by heart disease [4]. Despite advances in treatment developed before and after President Nixon’s declaration of the war on cancer, including surgery, radiation therapy, chemotherapy, and the more recent targeted therapies, only about 44% and 28% of cancer patients live more than 10 and 15 years, respectively, regardless of their quality of life [5].
Recently, several new therapeutic approaches have been developed that offer prospects for improving cancer prognoses, including immunomodulation and engineered redirected cellular therapies (Fig. 1). Data from clinical trial NCT00924326 showed that autologous anti-CD19 chimeric antigen receptor T cells (CART19s) were able to induce the regression of advanced follicular lymphoma in a patient. In parallel to the clinical outcome, B lineage cells were eradicated for at least 39 weeks post-treatment, which was attributed to CART19 therapy [6]. In 2011, our group at the University of Pennsylvania and our colleagues published two papers describing the use of autologous CART19 therapy to successfully treat patients with relapsed refractory chronic lymphocytic leukemia (R/R CLL) [7], [8]. These reports demonstrated for the first time that engineered T cells targeting CD19 malignancy could have durable therapeutic effects in humans.
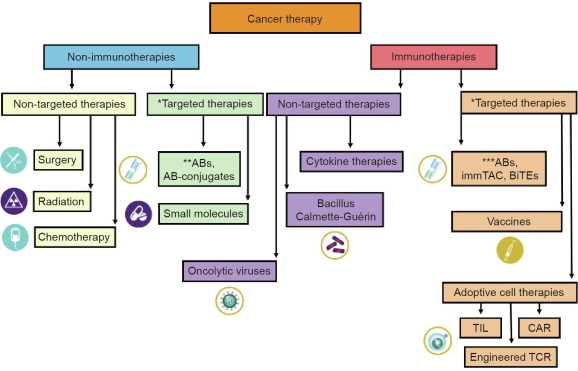
 Fig. 1. Classification of various cancer therapies based on the mechanism of action. *: therapeutic function derived by targeting a tumor-specific gene; **: monoclonal antibody (mAB) binding to cancer cell surface target and leading to cancer cell death, e.g., trastuzumab; ***: checkpoint inhibitors, e.g., pembrolizumab; AB: antibody; immTAC: immune mobilizing monoclonal T cellreceptors against cancer; BiTEs: bi-specific T cell engagers; TIL: tumor-infiltrating lymphocyte; CAR: chimeric antigen receptor; TCR: T cell receptor.
Fig. 1. Classification of various cancer therapies based on the mechanism of action. *: therapeutic function derived by targeting a tumor-specific gene; **: monoclonal antibody (mAB) binding to cancer cell surface target and leading to cancer cell death, e.g., trastuzumab; ***: checkpoint inhibitors, e.g., pembrolizumab; AB: antibody; immTAC: immune mobilizing monoclonal T cellreceptors against cancer; BiTEs: bi-specific T cell engagers; TIL: tumor-infiltrating lymphocyte; CAR: chimeric antigen receptor; TCR: T cell receptor.In order to clearly define and differentiate CART19 therapy from the other treatment modalities, Fig. 1 shows a schematic representation of current cancer therapies categorized based on the mechanism of action of drug targets. Cancer therapies can be grouped into non-immunotherapies and immunotherapies. Immunotherapies are treatments that directly modulate the patient’s own immune system in order to achieve beneficial clinical outcomes [9]. In contrast to immunotherapies, all other cancer therapies are grouped in the non-immunotherapy category. Oncolytic virus (OV) treatments possess characteristics of both groups in that they direct cell killing and induce antitumor immunity. Therefore, Kaufman et al. [10] have described OV treatments as a new class of immunotherapy drugs. Each group can be further classified into targeted therapies and non-targeted therapies. A targeted therapy is a treatment developed based on a cancer-specific target at the molecular level, which results in specific cancer cell killing due to target engagement [11], [12]. In contrast to targeted therapies, all others are in the non-targeted therapy group.
Under the category of targeted immunotherapies, there is a sub-group called adoptive cell therapy, which is comprised of ex vivo expanded unmodified tumor-infiltrating lymphocytes (TILs), engineered T cell receptor (TCR), and chimeric antigen receptor (CAR) T cells [13]. Adoptive T cell transfer therapy for cancer is intended to boost T cell activity in fighting cancer cells. CART19 therapy targeting CD19 consists of engineered autologous/allogeneic T cells that have been specifically directed to target the CD19 protein expressed on most of the B cells. These T cells have been “trained” to identify and kill B cells [8]. In a recent review of 20 published CAR T cell clinical trials, 11 were CART19 trials. Ten out of the 11 trials demonstrated some clinical benefits for patients with B cell malignancies [14]. Data from NCT02435849 have led the US Food and Drug Administration (FDA) to approve this therapy for the treatment of patients up to 25 years of age with B cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. It is now called Kymriah (tisagenlecleucel) [15]. Shortly thereafter, the FDA approved Yescarta (axicabtagene ciloleucel) to treat adult patients with certain types of large B cell lymphoma who have not responded to, or who have relapsed after, at least two other kinds of treatment based on data from NCT02348216 [16]. This was followed by the approval of Kymriah to treat large B cell lymphoma based on data from NCT02030834 [17]. The success of CART19 therapy has served as a proof of concept for engineered T cell therapies. It has also symbolized the beginning of a new frontier in modern medicine—engineered T cell therapies that can be directed to attack cancer cells or other disease-causing cells in order to treat cancer or other diseases [18], [19], [20].
CART19 therapy is essentially developed by following the “one gene, one drug, one disease” concept, regarding the creation of the CD19-specific CAR (CAR19) construct, which is the active pharmaceutical ingredient (API) (Fig. 2, Fig. 3) [21], [22]. However, it is unconventional in that the CAR19 needs to be packaged in attenuated lentivirus, or other types of vectors, and transduced into a patient’s own T cells [23], [24]. This is very different from conventional drug formulation, which incorporates the API with other inert ingredients. The goal of conventional drug formulation is to improve absorption, distribution, metabolism, and elimination (ADME) of the compound in order to maximize efficacy and minimize adverse effects (AEs) [25]. Because a CAR transgene is permanently integrated into the T cell genome, the equivalence to ADME is cell infusion, trafficking, proliferation, persistence, and apoptosis [26]. The journey that starts with collecting a patient’s own T cells and ends with the making of the final CAR T cell product involves many complicated steps and quality controls, which are difficult to automate. Establishing a manufacturing center that can produce the final clinical-grade CAR T cell products is as important as identifying the right drug target to build the optimized CAR for clinical development [27]. It is a process that is very new to the drug industry.
 Fig. 2. Drug development process and technological impact: fitting the engineered T cell therapy development process into the conventional drug discovery and development paradigm. Elements not currently utilized for T cell therapy are in grey italics. PK: pharmacokinetics; PD: pharmacodynamics; AEs: adverse effects; CK: cellular kinetics.
Fig. 2. Drug development process and technological impact: fitting the engineered T cell therapy development process into the conventional drug discovery and development paradigm. Elements not currently utilized for T cell therapy are in grey italics. PK: pharmacokinetics; PD: pharmacodynamics; AEs: adverse effects; CK: cellular kinetics. Fig. 3. Identification of API through lead optimization cycle: fitting the CARconstruct-perfection procedure into the lead optimization cycle. Elements not currently utilized for T cell therapy are in grey italics. ID; identification; TV: target validation; HTS: high-throughput screening; SAR: structure–activity relationship.
Fig. 3. Identification of API through lead optimization cycle: fitting the CARconstruct-perfection procedure into the lead optimization cycle. Elements not currently utilized for T cell therapy are in grey italics. ID; identification; TV: target validation; HTS: high-throughput screening; SAR: structure–activity relationship.This review is mainly focused on using CART19 as an example to explain how the preclinical and clinical development of CAR, the API for engineered T cell therapies, is similar to and different from the conventional drug discovery paradigm. We also emphasize the critical roles of biomarkers in guiding the drug development process. Furthermore, we discuss the impact of advanced technologies and computational tools in identifying better cancer-specific targets, novel CAR binding domains, and biomarkers that can be used to maximize clinical outcomes and minimize toxicity [28], [29].
2. Preclinical stage development of engineered T cell therapies
2.1. Target identification
Similar to conventional drug development, target identification is the first step in a gene-to-drug reverse pharmacology approach (Fig. 2, Fig. 3, Table 1) [30], [31]. A good target is the most crucial element in delivering the highest drug efficacy with the lowest incidence of AEs. There are general rules for the identification of the ideal drug target in the conventional drug development process. A drug target must be disease associated, which means that modulation of the target can result in a beneficial clinical outcome. It must be “druggable,” which means that the target can be modulated by an exogenous substance. The engagement of the target with an ideal corresponding substance will initiate a biological action that leads to high benefits and low AEs in a clinical setting. From a conventional standpoint, druggability mostly pertains to drug targets that can interact with small-molecule chemicals. With the successful development of monoclonal antibody therapies enabled by advancements in science and technology, however, it has become clear that druggability is really determined by technological availability [32], [33], [34].
Table 1. A comparison of the conventional drug discovery and development process with those of the CAR-based living drug.
| Development stages | Conventional process | CAR-based process |
|---|---|---|
| Target identification | Disease associated, druggable, and beneficial | Disease associated, druggable, and beneficial |
| Target class | Enzymes, receptors, and extracellular molecules | Extracellular molecules |
| Target validation | Druggability, specificity, disease association, and clinical benefit | Druggability, specificity, disease association, and clinical benefit |
| Lead identification & optimization | Hits, leads, and lead optimization | Rational design of CAR based on domain functions |
| Preclinical biological testing | In vitro activity, in vivo PK, and PD | In vitro activity, in vivo CK, and PD |
| Phase 0 | Testing proof-of-concept molecules | Not implemented |
| Phase I | Safety evaluation | Safety/efficacy |
| Phase II | Efficacy/safety | Efficacy/safety, Kaplan–Meier survival curve |
| Phase III | Benefit/risk | Not implemented |
| FDA review/Phase IV | Confirming safety and efficacy, post-marketing surveillance | Confirming safety and efficacy, post-marketing surveillance |
Regarding CAR T therapies, the domain structure of a CAR construct usually contains an ectodomain, transmembrane (TM) domain, and endodomain. The ectodomain contains antigen-binding and hinge regions, while the endodomain is composed of co-stimulation and activation regions (Fig. 4) [19], [35]. The binding site/specificity of a CAR construct is usually derived from the single-chain fragment variable (scFv) region of an antibody to the selected target that is accessible by the scFv, such as scFv region of an anti-human B cells antibody (FMC63) for CAR19 [36], [37]. Therefore, all extracellular molecules are druggable in the context of CARs as long as antibodies can be generated against these target antigens. An ideal cancer drug target for CAR is cancer-specific, extracellular, and interacting with an antibody. CARs have been engineered to target proteins, glycopeptides, and gangliosides on the cell surface [38], [39], [40]. Unlike CAR-based engineered T cell therapies, the engineered, “transgenic” TCR (tgTCR)-based approaches are not restricted to membrane antigens [41]. However, CAR-based therapies are independent of antigen processing or major histocompatibility complex (MHC) expression of the target cells [35], [42].
 Fig. 4. Major elements in one of the current CAR19 constructs. scFv: single-chain fragment variable; EF-1α: elongation factor-1α; hCD8α: human CD 8α; 4-1BB: human CD137 or tumor necrosis factor receptor superfamily member 9 (TNFRSF9); CD3ζ: T-cell surface glycoprotein CD3ζ chain or T-cell receptor T3ζ chain; VH: heavy chain variable region; VL: light chain variable region.
Fig. 4. Major elements in one of the current CAR19 constructs. scFv: single-chain fragment variable; EF-1α: elongation factor-1α; hCD8α: human CD 8α; 4-1BB: human CD137 or tumor necrosis factor receptor superfamily member 9 (TNFRSF9); CD3ζ: T-cell surface glycoprotein CD3ζ chain or T-cell receptor T3ζ chain; VH: heavy chain variable region; VL: light chain variable region.Cancer targets for engineered T cell therapies have been selected using the following criteria: ① neoantigens—that is, tumor-specific mutations, such as epidermal growth factor receptor variant 3 (EGFRvIII) [43], [44], [45]; ② proteins expressed on tumors and normal tissues, where the AEs resulting from the absence of the protein target in normal tissue is manageable, such as CD19 [46]; ③ differentially expressed proteins—that is, with high expression on tumors and very low expression on normal tissues, such as human epidermal growth factor receptor 2 (HER2) [47]; ④ cancer/testis (CT) antigens, such as NY-ESO-1, a cytoplasmic protein, which is processed and presented in the context of MHC class I for TCR recognition [48], [49]; and ⑤ viral oncogenes, such as HPV-16 E6 [50].
The most successful application of CAR T therapy thus far has been the redirecting of T cells to target CD19. CD19, a 95 kDa single TM glycoprotein, was known to be an ideal target to treat B cell malignancies since the 1980s [51]. Amino acid residues 1–273 form the extracellular region of CD19, which contains three cysteine disulfide bonds and several sites for glycosylation. Residues 274–298 are embedded in the cell membrane, while the rest of the 242 amino acids are in the cytoplasm [52]. In a complex with CD21 and CD81 on the cell membrane, CD19 functions as a co-receptor of the B cell receptor (BCR), which greatly sensitizes/potentiates BCR activity [53], [54].
CD19 is ubiquitously expressed on all stages of B cells except for hematopoietic stem cells (HSCs) and certain plasma cells. It is also expressed on most B lineage leukemia and lymphoma cells [51], [55]. It is expected that a successful adoptive CART19 transfer will eliminate B lineage malignancies and normal B cells leading to B cell aplasia. As the treatment does not eradicate the vital HSC, and as B cell aplasia is treatable with intravenous immunoglobulin replacement, CD19 is an excellent target for eliminating B lineage malignancies via the adoptive transfer of CAR T cells due to its antibody accessibility, tumor specificity, and non-lethal normal organ tissue distribution [46], [56].
2.2. Target validation
The conventional drug target validation process, in a gene-based drug development, has been very systemic in the pharmaceutical industry in the past decades. This process starts with evaluating the general information of a target, such as target class (i.e., receptor, enzyme, etc.), sequence homology among different species, gene family, and tissue/cell distribution. The disease association of a drug target is revealed by naturally occurring mutations, which can be the case for some targets, such as sclerostin [57]. Drug target disease association is proven by a genetic model, such as a knockout mouse model, which will exhibit beneficial or AEs in the absence of the drug target [30], [58]. Next, a beneficial therapeutic outcome needs to be proven when manipulating the drug target by increasing or decreasing its activity using candidate drugs in animal models. The drug target is further validated in clinical trials. The target validation process is synchronized with the development of the active pharmaceutical ingredient. FDA approval of a drug is the final validation of the corresponding drug target.
As an engineered T cell therapy exerts its clinical benefit by eliminating the target cells that a target gene-specific CAR recognizes, a target gene knockout model is not relevant for this modality of drug target validation. In addition, drug target selection is focused on molecules that are specific for the cells that need to be removed, such as cancer cells. Therefore, target validation for engineered T cell therapies is a process to prove that engineered T cells can selectively lyse the intended target cells in cell-based assays and eliminate/inhibit the target cell growth in a xenograft mouse model. For example, T cells transduced with various CAR19 constructs had been shown to lyse cells expressing CD19. CART19 cells were further proven to exhibit anti-B cell malignancy using an immunodeficient mouse xenograft model [37], [59].
2.3. Candidate drug development
After over 100 years of synthetic pharmaceutical development history, a routine procedure called the structure–activity relationship (SAR) drug lead optimization process has been established to select drug-like candidates for clinical trials (Fig. 3) [30]. Through extensive in vitro assay screening, selected compounds with desired biological activity and pharmacokinetic (PK) profiles are tested in animal models to further validate their in vivo PK and pharmacodynamics (PD). PD includes measurements of efficacy and AE derived from the tested compounds. It is a process that is heavily dependent on available technologies and on biomarkers that can be measured to reveal compound activities.
The current generation of CAR constructs have been engineered using rational design principles, which have yielded lead candidates for validation in cell-based assays and mouse models (Table 1) [37], [60], [61], [62]. It is also important to evaluate the post-infusion PK profile of engineered T cells in mouse models and in patients; however, as the drug is in the form of living cells, the term cellular kinetics (CK) is currently adopted in this context. As discussed in our recent study, many of the conventional PK analysis concepts such as maximum plasma drug concentration (Cmax), the area under the plasma concentration–time curve (AUC), and last measurable plasma concentration (Clast) can be usefully applied to CK [26], [37].
Even though engineered T cell therapies are only in the early stages of commercialization, the process from the selection of a target gene to the generation of a candidate drug is very systemic for the current reported targets. In the lead optimization and candidate drug selection phases, the assays and technological platforms that are implemented are the same as those used for target validation. The steps for candidate drug development are as follows: First, antibodies that recognize the selected drug target are identified, with the goal of obtaining the scFv sequence. Second, the CAR construct is made by inserting the scFv into an established CAR backbone. One of the CAR backbones used by our group contains 4-1BB/CD3ζ signaling domains for co-stimulation and activation functions, a CD8α hinge region, and TM domains (Fig. 4). The extracellular domains contain a CD8α leader and scFv specific for the drug target [37], [59], [60], [61], [62]. Third, the DNA sequence confirmation of the construct is performed before lentiviral packaging and transduction into activated human T cells; this is coupled with ex vivo cell expansion [27]. The activity of the final CAR T cells is studied extensively in preclinical models.
Milone et al. [37] have reported a detailed study on the optimization of CAR19 construct, which serves as a textbook example of the candidate drug/API optimization process for engineered T cell therapy. The final CAR19 construct was optimized for its expression promoter and intracellular co-stimulation/activation domains (Fig. 4). In in vitro studies, they first evaluated the effect of different promoters on gene expression in order to select the one that provides high and stable expression of the CAR19 in CD4 and CD8 T cells; they selected the elongation factor-1α (EF-1α) promoter. Second, they evaluated surface CD19 scFv expression via flow cytometry/Western blotting and ex vivocell expansion using constructs containing the EF-1α promoter and various co-stimulation/activation domains. These results confirmed the similar levels of expression of various CAR19s regulated by the same promoter, but did not differentiate the constructs containing different co-stimulation/activation domains in terms of activity, such as cytotoxicity or cytokine production. Third, the cell function of CART19s was evaluated through cytotoxicity assays. Incubation of CART19s with K562 wild type (Kwt) and K562 expressing CD19 (K19) demonstrated that CART19s containing 4-1BB/CD3ζ domains worked best in lysing K19, with minimal cytolytic activity in CD19-negative Kwt cells. This construct also effectively lysed human primary pre-B ALL cells expressing physiologic levels of CD19. Fourth, further profiling of the various CAR19 constructs was carried out by measuring cytokine production of the corresponding CART19. CART19 CD4+ and CD8+ T cells expressing different CAR19s were incubated with K19/Kwt, and levels of interleukin (IL)-2, interferon (IFN)-γ, IL-4, and IL-10 in cell supernatant were measured and analyzed. These results clearly demonstrated that different CAR19s had different activities of cytokine production. Fifth, they went on to evaluate T cell proliferation triggered by CART19 engaging with K19 cells. The difference in the co-stimulation/activation domains was clearly revealed in this assay, and CART19 cells containing 4-1BB/CD3ζ domains showed a CD19 antigen-independent proliferative capacity.
Further evaluation of CART19 transduced with different CAR19s was carried out using an immunodeficient mouse model of human pre-B ALL. The in vivo data again revealed the functional difference among various CART19 products and demonstrated the superior anti-leukemic efficacy in vivo of CART19 cells containing 4-1BB/CD3ζ domains. The in vitro and in vivo data on various CART19 products led to the ultimate selection of a CART19 patient-specific T cell product containing EF-1α_4-1BB/CD3ζ CAR19 construct as the candidate drug.
2.4. Historical preview of scFv and signal domain selection for CAR19
Several mouse anti-human CD19 monoclonal antibodies (αCD19s) had been described before the era of CART19 engineering. Uckun et al. [51] extensively characterized B43 monoclonal antibody (mAB), an αCD19 derived by immunizing BALB/c mice with tumor cells isolated from Burkitt’s lymphoma, and demonstrated that B43 recognized the same epitope as B4, AB1, BU12, F974A2, HD37, and SJ25-C1. In addition, clones FMC63, HIB19, and 4G7 are widely utilized antibodies to CD19 in clinical approaches and in research. B4 mAB was generated by immunizing a BALB/c mouse with B cell CLL (B-CLL) [63]. HD37 mAB was produced by immunizing BALB/c mice with cells from a hairy cell leukemia patient [55]. SJ25-C1 and 4G7 were derived by immunizing mice with NALM1 + NALM16 cells and B-CLL, respectively (BioLegend). FMC63 was derived by immunizing BALB/c mice with B-CLL cell line JVM3 [64]. Immunogen information could not be found for AB1, BU12, F974A2, or HIB19.
HIB19 is noted to partially block the binding of B43 (BD Biosciences), while FMC63 has been demonstrated to block the binding of HIB19 or SJ25C1 [65]. The existing data inevitably lead to the conclusion that all the αCD19s described above are interacting with the extracellular domain of CD19 in similar immunogenic areas. Another common aspect of these αCD19s is that the immunogens for the known ones are all an endogenous form of human CD19 on malignant B cells.
Major reported CART19 trials sponsored by Juno, Kite Pharama, Novartis, the Fred Hutchinson Cancer Research Center, and the Chinese PLA General Hospital all used murine anti-CD19 scFv—in the case of Juno, these were derived from SJ25-C1, whereas the rest were derived from FMC63 [64], [66]. Blinatumomab, a bi-specific T cell engager that was recently approved by the FDA, contains the CD19 scFv derived from HD37 [55], [67].
T cell, but not B cell, immune reactions against the murine scFv region have been reported for some patients in clinical trials. Efforts have been made to humanize the scFv region in order to reduce immunogenicity derived from the CAR19 transgene [68], [69].
Most CART19 trials have employed a second-generation CAR containing either CD28 or 4-1BB co-stimulatory domain [14], [66]. In their preclinical CART19 evaluation, Milone et al. [37] showed data in comparison with CD28 in order to demonstrate that 4-1BB had minimal induction of IL-4 and IL-10, CD19-independent CART19 proliferation, better engraftment/persistence in mice, and better antitumor efficacy. In ALL trials, both CD28 and 4-1BB performed similarly in regard to response rate and toxicities, albeit with subtle differences. Studies utilizing co-infusion of CD28 and 4-1BB CART19 may be able to delineate the properties of the two co-stimulatory domains [70].
3. Clinical stage development of engineered T cell therapies
3.1. Engineered T cell therapy clinical trial design
For conventional drug development, there is a systematic approach to test candidate drugs (Fig. 2, Table 1) [30]. Targeted cancer therapies are differentiated from the old chemotherapy/cytotoxicity. The clinical trial design for these agents emphasizes target engagement. Three levels of on-target pharmacodynamic endpoints are evaluated in a trial; these include target engagement for proof of mechanism (POM), phenotypic change for proof of principle (POP), and clinical outcome for proof of concept (POC) [71]. In the past two decades, drug companies have been trying to improve the success rate and reduce the cost and time of making new drugs. Phase 0 trials are implemented for imperfect candidate drugs—proof-of-concept molecules (POCM)—in order to gain clinical insights and make quick Go/No-go decisions [72]. This new discipline has emerged to become what is known as exploratory medicine, experimental medicine, translational medicine, or discovery medicine. This discipline is an effort to bridge the gap between preclinical animal studies and clinical trials, as many of the animal models fail to predict clinical outcomes. The primary goal for Phase 0 can be to evaluate the PK/PD of a candidate drug through microdosing, to rank several non-perfect candidate drugs, and/or to validate the POM and POP of candidate drugs via biomarkers [71].
The primary goal of a Phase I clinical trial is safety evaluation and determination of the maximum tolerated dose of a drug candidate. In this setting, the PK profile, toxicity, and PD data will be documented. Usually, a Phase I trial involves 20–100 healthy volunteers for less than one year of study. The primary goal for Phase II is to evaluate drug efficacy and perform further safety evaluation. The PK profile, toxicity, and PD data are further documented. This study enrolls 20–300 patients, and lasts up to two years. The primary goal for Phase III is to assess additional information about the effectiveness in terms of clinical outcomes and overall risk–benefit evaluation. The PK profile, toxicity, and PD data are further documented. This study enrolls 300–3000 demographically diverse patients, and lasts up to several years. Phase IV studies are post-marketing surveillance trials. In these, the primary goals are ongoing safety monitoring and identifying additional uses of the drug [73].
The clinical trial design for CART19 therapy is a modified version of the paradigm described above (Table 1). As the drug is the last resort for many of the patients with relapsed refractory CD19+ B cell malignancies, and as CART19 was a promising experimental drug before 2017, the trial design is a combination of the conventional trials described above and expanded access [74]. The published Phase I studies, such as NCT01626495 and NCT01029366, were all carried out with terminally ill patients. In addition to evaluating the primary objectives of safety and feasibility of the treatment, many secondary objectives were included in order to prove the POM, POP, and POC and to study CART19 CK, differentiation, and host immunogenicity [8], [75]. The Phase II study NCT02435849 (ELIANA) of NCT01626495 had the primary objective of efficacy as measured by overall remission rate (ORR). The secondary objectives were extensive, with the goal of gaining further insights into the efficacy of this therapy. The impressive clinical benefit of CART19, as demonstrated by ELIANA, led the FDA to approve this drug, which is marketed as Kymriah [15].
3.2. A systemic biomarker-guided procedure in evaluating engineered T cell therapies in the clinic
Despite the fact that the pharmaceutical industry is a newcomer in developing CAR T cell therapies, academic researchers have established a systematic pipeline for the clinical development of engineered T cell therapies. Extensive details have been reported on evaluating the POM, POP, and POC in patients for PD evaluation; the persistence of CART19s for CK; and cytokine-release syndrome (CRS)/on-target off tissue for AE/safety. This research is summarized below [8], [26], [75], [76], [77].
Methods to evaluate POM. The engagement of CAR19 with CDR19 on the target cell surface is evaluated by measuring biomarkers in serum that are released when CART19s interact with CD19+ B cells. A panel of 30 analytes containing cytokines, chemokines (30-plex), and others was measured in patient serum and bone marrow (BM) samples collected at different time points post-treatment [8]. Elevation of analytes, such as IL-6, IFN-γ, and IL-8, is a good indication of target engagement. These analytes can be measured using commercial Luminex bead-based assays or enzyme-linked immunosorbent assays (ELISAs). Furthermore, in vivo expansion of CART19 cells is another parameter that demonstrates target engagement activity. The expansion of CAR19+ cells can be determined via flow cytometry by gating on CD3+ and CAR19+ cells, or through quantitative polymerase chain reaction (qPCR) on peripheral blood genomic DNA using primer pairs that are specific for CAR19 construct [8].
Methods to evaluate POP. Assays to study phenotypic change include the absolute lymphocyte count (ALC), CD19+ B cell enumeration via flow cytometry by gating on CD19+ and CD20+ B cells, or more complicated BM staining and computerized tomography scan imaging. The reduction of CD19+ B cells is a clear evidence of POP.
Methods to evaluate POC. These methods evaluate the clinical benefit on the patients after achieving the POM and POP. Results regarding tumor burden elimination, event-free survival, and feeling healthier post-treatment are all evidence to support the POC. This is not a redundant evaluation, as POC is not achieved for patients who develop CD19− B cell leukemia while POM and POP are achieved.
Methods to evaluate CAR19+ cell CK. CK evaluation post-treatment over time is more relevant for engineered T cell therapies than PK study. CAR19+ cells can be determined at different time points in serum, BM, or cerebral spinal fluid (CSF) samples via flow cytometry by gating on CD3+ and CAR19+ cells. The presence of CAR19 construct can also be quantitated over time through qPCR or digital PCR on genomic DNA (gDNA) extracted from peripheral blood or other tissues and expressed as copies per microgram of gDNA. Both flow and qPCR data correlate with each other, while the PCR method can be more sensitive. Similar parameters to PK can be deduced from the above measurements, such as the AUC, half-life (T1/2), and so forth [26].
Methods to evaluate persisting CART19 cell function. The effector function of the persisting CART19s can be confirmed through a degranulation assay using CD107α as a marker [78]. For the degranulation assay, CART19s at different post-infusion time points were collected and incubated with target cells. Cells were then analyzed through flow cytometry using degranulation marker CD107α paired with CAR19 marker [8]. An alternative method is to measure the IFN-γ level in the co-culture cell supernatant, which also evaluates CART19 function [37], [62].
Methods to evaluate AE. CRS is a major on-target AE in this modality of treatment. The aforementioned 30-plex assay, or other multiplex assay panels, can be used to monitor post-treatment effects, such as elevation of IL-6, IFN-γ, and IL-8. Other clinical symptoms include fever, myalgia, and nausea. For some patients, intensive care is required due to hypotension, capillary leak, and hypoxia. Macrophage activation syndrome may occur concurrently with CRS, as evidenced by high levels of ferritin, C-reactive protein, and soluble IL-2 receptor [75], [76], [77].
B cell aplasia is an on-target non-lethal AE in this treatment; it is an indicator of beneficial clinical outcome and can be managed by intravenous immunoglobulin supplementation [46].
Immunogenicity of the CAR19 is a concern for this category of drug. However, the presence of CAR19 cells several months post-infusion in multiple studies has suggested the absence of immunogenicity [8], [15], [26], [76]. Nevertheless, Turtle et al. [68] have reported a T cell immune response to CAR19 in some patients. The absence of humoral responses is not surprising, as the treatment eradicates B cells. However, the absence of cellular rejection of CAR19-containing murine scFv and novel junctional regions derived from the chimeric protein in most patients could be attributed to cyclophosphamide/fludarabine lymphodepletion treatment [79].
Neurologic adverse events were reported in several of the CART19 trials. In the NCT02435849 trial, 40% of the patients experienced some neurologic issues within eight weeks after infusion [15]. In the NCT02348216 trial, 64% of the patients had neurologic symptoms, of which 28% were grade 3 and higher [16]. Juno’s ROCKet trial was put on hold due to patients’ deaths from cerebral edema [80]. The risk factors for neurotoxicity, such as endothelial activation and increased blood–brain barrier, were reported by Gust et al. [81].
4. Technological impact on engineered T cell therapies
The CART19 triumph only marks the beginning of engineered T cell therapy industry. We anticipate the growth and maturation of this industry, enabled by continually advancing technologies and artificial intelligence. The following are some examples of areas that are being enhanced by technologies and computer-assisted analysis tools.
4.1. Expanding novel cancer-specific targets via genomics and bioinformatics tools
In the post-genomic era, advanced genomic technologies and bioinformatics will inevitably have an impact on tumor-specific target identification. A mass spectrometry-based multiplex protein target analysis method to identify cancer-specific mutant proteins has been proven feasible; this method can be utilized to identify cancer biomarkers for diagnostic kits development or novel drug targeting for cancer therapy [82]. Neoantigens are primarily being identified through whole-exome sequencing followed by bioinformatics analysis. Furthermore, when the method is coupled with TIL screening through the co-culture of polyclonal TIL population and antigen-presenting cells transfected with tandem minigenes derived from neoantigens, along with parallel single-cell RNA-seq, neoantigen-specific TCRs and the corresponding neoantigen can be identified [83], [84]. Through deep mining of multiple public transcriptomics databases, novel genes with a CT expression pattern have been identified in 19 cancer types [85].
4.2. Novel CAR binding domain identification: HTS on combinatorial antibody libraries
Although currently reported CARs, such as CAR19, were built based on existing antibodies, high-throughput screening (HTS) and lead optimization process are expected to play a critical role in future engineered T cell therapy development (Fig. 3) [37], [86]. The screening concept and technology for small-molecule drug discovery can be readily adopted to identify the ideal CAR binding domains [87]. Just as the combinatorial compound libraries have provided novel chemical platforms for small-molecule candidate drug identification, combinatorial fully human antibody libraries will be sources for mining scFv containing new binding sites or different binding affinity, which can be used to build CARs with desired specificity or affinity [88], [89], [90]. HTS-associated technologies and computational tools are expected to play important roles in the expansion of the engineered T cell therapy portfolio.
4.3. Discovery of biomarkers to predict the onset of CRS and assist management plans
CART19 treatment associated CRS can range from mild to severe, and the onset time varies. The ability to predict treatment-induced CRS as early as possible will improve beneficial clinical outcomes and reduce AE. In a retrospective analysis of accumulated patient samples by means of Luminex bead-based multiplex assays on 30 cytokines/chemokines and 14 soluble cytokine receptors, several predicative biomarker signatures were identified. For example, a three-variable regression model using IFN-γ, soluble gp 130 (sgp130), and IL-1 receptor antagonist (IL-1RA) results from the first 3 d post-infusion accurately predicted which patients developed severe CRS in the adult and pediatric cohorts. A decision tree model using sgp130, monocyte chemoattractant protein-1 (MCP1), and eotaxin also predicted the onset of CRS in patients [77]. In addition, a CRS grading scheme and treatment plan was developed and implemented for CRS management by University of Pennsylvania and Children’s Hospital of Philadelphia physicians, which can optimize efficacy and minimize toxicity [91].