1. Introduction
Minerals flotation exploits the difference in mineral surface properties in order to separate them via physico-chemical processes. To enhance such surface properties, different chemicals can be used that can be broadly classified as either: collectors, modifiers, depressants, or frothers. Lime (in the form of calcium oxide, CaO, or calcium hydroxide, Ca(OH)2) is a very common modifying reagent. Modifying reagents react either with the mineral surfaces or with collectors and other ions in the flotation pulp, resulting in a modified and controlled flotation response (Bulatovic, 2007a). More specifically, lime is categorized as an inorganic modifier, with its main role being modification of the pulp pH. The flotation response of sulphide minerals and their interactions with collectors (xanthates, dithiophosphates, thionocarbamates, and their derivates) is strongly pH dependent. Selective flotation, of one mineral over another, can thereby be achieved by varying the pulp pH. For example, chalcopyrite can be selectively floated from iron sulphides (pyrite, pyrrhotite) in a highly alkaline environment (pH > 11.5), while at lower pH values pyrite readily adsorbs collectors and reports to the flotation concentrate (Wang and Forssberg, 1991).
Alkalinity in sulphide flotation is generally controlled by the addition of lime. Alternatives to lime for pH control include soda ash (Na2CO3) and caustic soda(NaOH). Soda ash and caustic soda are, however, more expensive than lime. The former is only used to control the pH up to about 10.5, due to the highly buffered pH response of carbonate, while the second is a strong base that can increase the pH to 14. But it is highly corrosive, which can raise safety concerns, and its industrial use is mainly limited to the processing of non-metallic minerals (Wills and Napier-Munn, 2005, Bulatovic, 2007a).
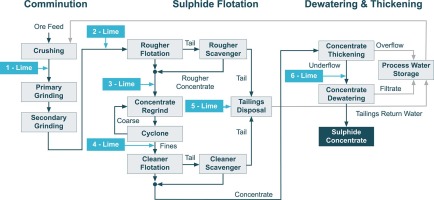
Sulphide ores are the major source of valuable metals such as copper, lead, zinc, and nickel, as well as gold, silver, and platinum group metals (PGMs), and flotation is the dominant industrial process to recover these metals via the production of a sulphide concentrate. Of total world copper-mine production, almost 80% originates from flotation of the copper (Cu) sulphides (Table 1). The ready availability and relatively low cost of lime makes it the preferred pH modifier used in the flotation of these ores. Dosage and addition points of lime in the circuit require however careful consideration for a cost-effective use of lime. This paper reviews the preparation of lime and its main mechanisms of action in the flotation of sulphide minerals, as well as the major industrial applications.
Table 1. Annual world mine production of copper by industrial process (2013 data from Matos et al., 2015).
| Mine Production (metric tons) | Percentage (%) | |||
|---|---|---|---|---|
| Flotation | 12,700,000 | 78.9 | ||
| Leaching, electrowon | 3,470,000 | 21.6 | ||
| Total | 16,100,000 | 100.4 | ||
2. Lime formulation, synthesis, and production
2.1. Calcination
Quicklime (CaO) is produced from the calcination of limestone (CaCO3), a sedimentary rock of mainly marine sediments containing calcium carbonates in a range of crystal forms. The chemistry and the physical properties of the originating limestone significantly affect the calcination process and hence the final lime product qualities and performance. During calcination calcium carbonate undergoes thermal decomposition when heated at temperatures higher than 800 °C. The products of this decomposition are calcium oxide (quicklime) and carbon dioxide gas:CaCO3 + heat = CaO + CO2ΔH(900 °C) = 2953 kJ/kg CaO; ΔG(900 °C) = −35 kJ/kg CaO
The reaction is highly endothermic, thus requiring an energy fuel source such as coal or gas (Herrier et al., 2010).
The decarbonation process of expelling CO2 from calcium carbonate happens on a migrating front starting on the limestone surface within the calcination kiln. The exterior of the stone decarbonates first, due to the fact that limestone and lime are poor heat conductors. The heat then penetrates deeper into the stone enabling further thermal decomposition (core-shell model). The purpose of decarbonation is to increase the conversion extent of CaCO3 into CaO. The driving force of this process is the equilibrium between the carbonate and the partial pressure of carbon dioxide. At atmospheric pressure, the temperature must exceed 900 °C to drive the reaction towards CO2 evolution (Boynton, 1980). The extent of conversion can be increased by calcination intensity (temperature) or duration. However, the requirement to achieve a high level of conversion is practically limited by the risk of over-burning when calcination conditions are too severe. Over-burning reduces the reactivity of lime when reacting in aqueous mineral slurries and thereby the utilization efficiency of the reaction.
2.2. Quicklime porosity
The CO2 gas expelled from the limestone during calcination causes porosity within the resulting quicklime CaO product. Theoretically, the porosity could be as high as 54%. In practice, however, lower porosity is observed due to sintering effects of CaO grains, as illustrated in Fig. 1. Increased calcination temperatures increase this effect. The presence of different impurities such as iron, sodium, potassium, phosphate, chlorides, or sodium can also promote the formation of larger grains via interstitial liquid phases (Boynton, 1980) even at lower temperatures. Larger CaO grains will have an important effect on the quicklime properties, with lower surface area (from larger grains) lowering the reactivity of quicklime with water, thus affecting the resulting milk of lime quality and utilization efficiency.
 Fig. 1. SEM images and diagram of lime grain sintering as a function of the temperature, internal Lhoist.
Fig. 1. SEM images and diagram of lime grain sintering as a function of the temperature, internal Lhoist.2.3. Calcination fuel source effects
The fuel source is an important driver of quicklime cost and quality. Fuel cost generally represents approximately 50% of the final quicklime production cost (Boynton, 1980). Fuel sources include coal, natural gas, oil, plastic waste, or biomass (Oates, 1998). Two key fuel selection criteria are cost-per-GJ and level of impurities. Impurity levels are important because all non-carbon-based impurities will report to the final quicklime product. Coke and heavy fuel oil, for example, contain high levels of sulphur (Oates, 1998). High sulphur content in the calcination fuel has a coarsening effect on Ca(OH)2 particles during the slaking process that, in turn, could result in poor lime utilization efficiencies during mineral processing (Ineich et al., 2017).
2.4. Quicklime slaking
Adding dry quicklime to plant circuits is not generally practiced, because of problematic operating issues such as dusting, clogging and poor dose controllability (National Lime Association, 1995). Instead, quicklime is generally slaked before addition to the mineral processing circuit as a slaked lime slurry, also known as milk of lime, composed of suspended hydrated calcium hydroxide Ca(OH)2 at a solids concentrations in the range 15–25% w/w. CaO particles exothermically react with water to form Ca(OH)2.CaO + H2O = Ca(OH)2 + HeatΔH(50 °C) = −1175 kJ/kg CaO; ΔG(50 °C) = −1020 kJ/kg CaO
These Ca(OH)2 particles are smaller in size than the CaO particles because of capillary water penetration into the porous CaO and subsequent decrepitation of the CaO upon hydration to form Ca(OH)2. The extent of water penetration, lime hydration and subsequent decrepitation is dependent upon the CaO porosity, which, in turn, is dependent upon the calcination conditions as indicated earlier. A low porosity CaO will result in coarser Ca(OH)2 particles, causing reduced lime utilization efficiency.
2.5. Ca(OH)2 particle size effects
It is important to note that Ca(OH)2 particles generated in the slaking process are only slightly soluble in water (about 1.4 g.l−1 CaOH2 at ambient temperature, (National Lime Association, 1995) and that it is the soluble calcium (Ca2+) and hydroxyl ions (OH−), rather than the solid Ca(OH)2, that participate in target reactions (Giles et al., 1993). Dissolution of Ca2+ and OH− from the surface of solid Ca(OH)2 particles is the limiting step in the reaction. Finer Ca(OH)2particles, with a large surface area of dissolution, result in improved kinetics and utilization efficiency, compared to larger Ca(OH)2 particles. The preferable Ca(OH)2 particle size distribution is a d50 lower than 15 µm. The particle size of the slaked Ca(OH)2 is therefore an important determining factor influencing overall reagent utilization efficiency and kinetics. Fine particle size is also important to avoid particle coating effects and settling problems in lime distribution pipes (Hassibi and Singh, 2012). Ca(OH)2 particle size is mainly determined by the following factors:
-
•
Reactivity of the lime (influenced by the calcination process conditions and fuel), with more reactive lime typically resulting in finer Ca(OH)2 particles (National Lime Association, 1995).
-
•
Soluble elements contained in the slaking water, e.g. high concentrations (>500 mg/L) of sulphate in the water will coarsen the milk of lime (Potgieter et al., 2003).
-
•
Operating temperature of the slaker, i.e. higher slaking temperatures (65–85 °C) generally result in finer Ca(OH)2 particles. (Zinck et al., 2000) This phenomenon is particularly relevant for low reactivity lime, but less relevant for higher reactivity lime. For a continuous production process, the slaking temperature is primarily controlled by the lime-to-water feed ratio of the slaker (typically at mass ratio of 1:6 to 1:4) and the reactivity of the lime.
-
•
Extent of mechanical abrasion or comminution during or after slaking. Some slakers, for example, make use of comminution methods (as in ball mill slakers) to reduce the particle size of the slaked lime. This is particularly used in scenarios where other factors would otherwise result in coarse Ca(OH)2 (Gartner and Diaz Chavez, 2015).
2.6. Grits management
During the slaking reaction the impurities and unburned carbonate in the lime will not be slaked. Unreacted and inert particles that do no convert to Ca(OH)2, known as grits, are, in some plants, rejected from the slaked lime product prior to distribution into the plant circuit (National Lime Association, 1995). Grits rejection is, however, not required for scenarios where grits particles are sufficiently small and their introduction into the mineral processing circuit is negligible relative to the ore feed.
2.7. Quicklime quality and reactivity metrics
Lime quality is commonly assessed by measuring the available CaO content (EN 459-2 or ASTM C110). This metric, however, does not give an indication of reaction kinetics with water and has been found to be an insufficient predictor of utilization efficiency (Ineich et al., 2017). Determining the kinetics of the slaking reaction consists of tracking the temperature rise of water when quicklime is added to it, with a rapid rise in temperature indicating high reactivity. A T60, (EN 459-2) corresponds to the time required for 600 g of water at 20 °C to reach 60 °C after addition of 150 g of quicklime. T60 values typically range from 0.5 to 5 min. A T60 lower than one minute may present safety concerns due to the risk of boiling, while a T60 > 5 min is an indicator of poor slaking behavior, again increasing the potential for larger particles of Ca(OH)2 in the final product and resulting in poor utilization efficiency (Ineich et al., 2017).
2.8. Safety considerations
Lime is not classified as acutely toxic. Nonetheless, it is an irritant for respiratory tracks (H335) and skin (H315), and it is corrosive for the eyes (H318) (Lime SDS, 2019). Specific care and personal protective equipment are required to avoid contact with skin and eyes as well as inhalation and ingestion. This is particularly relevant due to the exothermic reaction of lime with water from moist skin, eyes and internal mucous membranes. Care should also be taken to prevent water ingress into lime during transport and storage. In severe cases, the heat released from the hydration reaction may ignite plastic liners (big bags) used during transport and storage. Appropriate firefighting measures, using dry powder foam or CO2, are required for such scenarios as water is not a suitable extinguishing media (Lime SDS, 2019).
3. Lime interactions with collectors and depressants
Flotation is an electrochemical process, in which mineral surfaces react with reagents in solution to form hydrophobic complexes that impart flotability to the mineral particles. In sulphide minerals flotation, these reactions involve oxidation of some species and reduction of others, resulting in selective adsorption of collector on the mineral surfaces (Chander, 1988).
The collectors used in sulphide flotation are anionic collectors (xanthates, dithiophosphates, thionocarbamates, etc.), characterized by the presence in the molecule of either sulphydryl (SH) or thiol (C-SH or R-SH) groups. Collector decomposition and the formation of species at the mineral surface is a complex process that is both kinetically and thermodynamically controlled (McFadzean et al., 2015). Different theories were developed for the mechanisms of collector adsorption (Gaudin, 1957, Fuerstenau et al., 1968, Allison et al., 1972, Wang and Forssberg, 1990, Wang and Forssberg, 1991, Miller et al., 2002). It is now accepted that flotation occurs due to the adsorption of either the negatively charged thiol collector (with the formation of a hydrophobic metal-collector species at the mineral surface) or of a neutral molecule or dimer of the collector (Fairthorne et al., 1997a, Fairthorne et al., 1997b, Fuerstenau, 2005) (Fig. 2).
 Fig. 2. Mechanisms for sulphide mineral surface/collector interaction. Both oxydryl and sulphydryl ions interact with the mineral surface, and the two compete for adsorption. Adapted from Fuerstenau (2005).
Fig. 2. Mechanisms for sulphide mineral surface/collector interaction. Both oxydryl and sulphydryl ions interact with the mineral surface, and the two compete for adsorption. Adapted from Fuerstenau (2005).Lime, mainly added as slaked Ca(OH)2 as a pH modifier, plays an important role in the selective flotation and depression of sulphide minerals. Sulphide flotation is generally conducted in alkaline conditions, which is where the collectors are more stable. Early literature suggested that in a mixed minerals/collector system there was an optimum pH range in which each mineral could be floated. Wark and Cox (1934) showed that the flotability of each sulphide mineral was proportional to the relative concentration of collector and hydroxyl ions [DTP−]/[OH−] and calculated a critical pH value for flotation of chalcopyrite, galena and pyrite with dithiophosphate (Fig. 3).
 Fig. 3. Relationship between concentration of sodium diethyldithiophosphate and critical pH value for the flotation of chalcopyrite, pyrite and galena. Curves at constant [DTP−]/[OH−]. Flotation occurs under conditions to the left of each curve but does not occur to the right of the curve. Adapted from Wark and Cox (1934).
Fig. 3. Relationship between concentration of sodium diethyldithiophosphate and critical pH value for the flotation of chalcopyrite, pyrite and galena. Curves at constant [DTP−]/[OH−]. Flotation occurs under conditions to the left of each curve but does not occur to the right of the curve. Adapted from Wark and Cox (1934).The pulp redox potential, which is correlated to pulp pH and dissolved oxygen by the relation:(1)also plays a role in the oxidative reactions at the surface of minerals and the complexation with collectors. Richardson and Walker (1985) showed that the flotation of chalcocite, bornite, chalcopyrite and pyrite with potassium ethyl xanthate is controlled by the conditioning redox potential (Eh). The active surface entities were found to be metal xanthates for chalcocite and bornite, dixanthogen for pyrite, metal xanthate for initial flotation of chalcopyrite and dixanthogen for full flotation. For pyrite, Fuerstenau et al. (1968) found a window for flotation between pH 3 and pH 10 in the presence of ethyl xanthate. The upper limit of pH 10 is actually explained as the limit for oxidation of xanthate to dixanthogen on the surface of pyrite (+163 mV). Above pH 10, this oxidation does not occur.
The different pH/Eh ranges of thermodynamic stability of the metal-collector species allows for the selective flotation of metal sulphides, by conditioning both pH and redox potential. Lotter et al. (2016) overlapped the results of previous flotation studies on a number of sulphide minerals with the Pourbaix stability diagrams of both xanthate and dithiophosphate species in water solution, identifying regions in the pH-Eh domain in which collector adsorption is more or less likely on each mineral, and selective flotation can, in principle, be achieved (Fig. 4). In a flotation plant, lime can be used to control the pulp pH and NaSH/SO2 to regulate the Eh in the proper range for selective flotation to occur. From a practical point of view, however, it is difficult to maintain the pH/Eh at the specified range in flotation if air is used as the carrier. Indeed the dissolved O2 will continuously shift the potential towards higher values. In such conditions, improved Eh control may be achieved by using N2 as carrier flotation gas, instead of air.
 Fig. 4. Empirically arranged flotation domains of chalcopyrite (Cpy), bornite(Bor), chalcocite (Cc), and Covellite (Cov) in terms of Eh and pH values, with Pourbaix Diagram of butyl xanthate and ethyl dithiophosphate overlain. Adapted from Lotter et al. (2016).
Fig. 4. Empirically arranged flotation domains of chalcopyrite (Cpy), bornite(Bor), chalcocite (Cc), and Covellite (Cov) in terms of Eh and pH values, with Pourbaix Diagram of butyl xanthate and ethyl dithiophosphate overlain. Adapted from Lotter et al. (2016).The mechanisms for collector adsorption in mixed sulphide mineral systems are also influenced by electrochemical interactions between minerals, and between minerals and grinding media. Sulphide minerals are semiconductors with distinct rest potentials (Table 2), and when two sulphide minerals come into contact there is a transfer of electrons from the less cathodic to the more cathodic one (Yelloji Rao and Natarajan, 1989, Rao, 2004, Nava and Gonzalez, 2006, McFadzean et al., 2015, Fallon et al., 2017).
Table 2. Rest potential of selected sulphide minerals at pH 4 and 7. Adapted from Fallon et al. (2017).
| Mineral | Formula | Rest Potential at pH 4 (pH 7) |
|---|---|---|
| Pyrite | FeS2 | 0.66 (0.45) |
| Marcasite | FeS2 | 0.63 |
| Chalcopyrite | CuFeS2 | 0.56 (0.34) |
| Sphalerite | (Zn,Fe)S | 0.46 |
| Covellite | CuS2 | 0.42 |
| Bornite | Cu5FeS4 | 0.40 |
| Galena | PbS | 0.28 (0.17) |
| Argentite | Ag2S | 0.28 |
| Stibnite | Sb2S3 | 0.12 |
| Molybdenite | MoS2 | 0.11 |
In grinding mills, fresh and reactive mineral surfaces are continuously generated. Galvanic surface reactions between minerals and grinding media can be intense (forged steel grinding media is reactive in contact with the sulphides), causing either the formation of hydrophobic sulphur species upon oxidation, or dissolution of metal ions (Cu2+, Pb2+) which can then activate other minerals (Ball and Rickard, 1976, Leppinen, 1990, Senior and Trahar, 1991, He et al., 2005, Bruckard et al., 2011, Moslemi et al., 2012). As a consequence, mineral surfaces are contaminated (i.e. activated), and contamination may cause a shift in the pH-Eh flotation domain. For example, chalcopyrite (CuFeS2) in an ore can release Cu and Fe ions in solution upon galvanic contact with pyrite (Fig. 5).
 Fig. 5. Mechanisms of galvanic interactions between sulphide minerals in solution. Adapted from Fallon et al. (2017).
Fig. 5. Mechanisms of galvanic interactions between sulphide minerals in solution. Adapted from Fallon et al. (2017).Metal ions in solution may then precipitate in the form of hydroxide, but copper can also be adsorbed on the surface of pyrite, making the mineral readily flotable (Fig. 6) with the usual thiol collectors (Fornasiero and He, 2008). Lime can be added to depress pyrite, with both the hydroxyl and calcium ions contributing to the formation of hydrophilic precipitates on the pyrite surface, but higher flotation pH is needed when pyrite is copper activated (pH > 11.5), thereby imposing a high lime consumption. Every ore is, however, different and it is difficult to determine a priori lime consumption for a specific ore. Also, lime demand in the plant can change significantly over time due to feed and process-water chemistry variability.
 Fig. 6. Proposed effect of copper activation on pyrite flotation.
Fig. 6. Proposed effect of copper activation on pyrite flotation.3.1. Interactions between lime and depressants
Lime in flotation, as a pH modifier, can also regulate not only collector adsorption, but also the effect of other commonly-used depressants such as sodium sulphide or sodium cyanide (Yoon, 1981, Ghiani et al., 1984, Wang and Forssberg, 1996). Cyanide acts by dissolving copper from the surface of the activated sulphides, thus preventing collector adsorption. Lime is used in combination with sodium cyanide for selective flotation of multi-metallic ores, by regulating the effective concentration of cyanide needed (Wark and Cox, 1934) (Fig. 7). For example, this strategy can be applied to pyrite. According to Fig. 7 pyrite could be separated from chalcopyrite at pH 7.5 in the presence of NaCN (30 mg/L). Separation may also occur in the absence of cyanide, but only in a narrow pH window between 10.5 and 11.
 Fig. 7. Relationship between concentration of sodium cyanide and critical pH value for the flotation of different sulphide minerals (at 25 mg/L potassium ethyl-xanthate). Flotation occurs under conditions to the left of each curve but does not occur to the right of the curve. Adapted from Wark and Cox (1934).
Fig. 7. Relationship between concentration of sodium cyanide and critical pH value for the flotation of different sulphide minerals (at 25 mg/L potassium ethyl-xanthate). Flotation occurs under conditions to the left of each curve but does not occur to the right of the curve. Adapted from Wark and Cox (1934).4. Calcium ion interactions with mineral surfaces
The role of lime in flotation is not limited to pH modifier, indeed the hydroxyl ion (OH−) is not the only active component of lime. Calcium ions, which are released in solution, can also interact with the mineral surfaces, producing different effects in flotation, either favorable or detrimental to the process.
Lime was found to be a preferred pyrite depressant over other pH regulators such as sodium hydroxide or soda ash. Several studies highlighted the specific role of calcium ions in the depression of pyrite. Tsai et al., (1971) observed that the adsorption of Ca2+ ions increases rapidly on pyrite as the alkalinity of solutions increases, and attributed depression to the adsorption of Ca2+ ions associated with OH− ions. Hu et al. (2000) attributed the depression of pyrite by lime to the surface formation of Ca(OH)2, CaSO4 and Fe(OH)3, as determined by XPS analysis (Fig. 8). Niu et al. (2018) also showed that the poor flotation response of pyrite at high pH was due to the prevailing adsorption of CaOH+species on the surface, indicating a stronger affinity of the hydrophilic calcium species towards pyrite with respect to the collector (Fig. 9). Calcium clearly plays a specific role. All alkali do not exhibit the same behavior during flotation of different sulphide minerals.