1. Forkhead box P3 as a “master regulator” of regulatory T cells
Regulatory T cells (Tregs) are the dominant subset of T cells that suppress host immunity. They were first described as a cluster of differentiation (CD)4+CD25high T cell population [1], and were later found to dominantly express the forkhead box P3 (FOXP3) protein [2]. The history of FOXP3 has been well summarized in a review written by Ramsdell and Ziegler [3]. Since the identification of Tregs, an enormous number of studies have been conducted to reveal the role and function of this unique T cell subset.
FOXP3 is a transcription factor belonging to the forkhead box family of transcription factors. It contains three major domains: the N-terminal domain, which is called the “repressor domain” [4]; the zinc finger and leucine zipperdomain (ZL domain), which is centrally located and facilitates the formation of FOXP3 homo-dimers, or heterodimers with other FOXP family members such as FOXP1 and FOXP4 [5]; and the highly conserved forkhead (FKH) domain at the C terminus, which is responsible for DNA binding.
The Foxp3 gene has been cloned as a causal gene for scurfy mice [6] and for immunodysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome in humans [7], [8]. Scurfy mice have 2 base pairs (bp) insertions in the exon 8 of Foxp3, which results in malfunction of the FOXP3 protein and the death of mice at approximately three weeks of age [6]. On the other hand, more than 70 FOXP3 mutations have been found in IPEX patients thus far [9], [10], [11]. The majority of these mutations are located in the FKH domain, but they are also found throughout the entire protein, indicating that all of the domains in FOXP3 could be important for its immune-suppressing function.
FOXP3 is known to form large complexes involving many proteins. Li and Greene [12] were the first to discover that FOXP3 forms large complexes ranging in size from 300 kDa to more than 1200 kDa. Later, using biochemical and mass spectrometric approaches, Rudra et al. [13] found that FOXP3 is able to form complexes with more than 360 proteins. Interestingly, Kwon et al. [14] have shown that FOXP3 forms different sizes of complexes, and that each complex has distinct cofactors. For example, FOXP3 forms large complexes with RelA, lysine acetyltransferase (KAT)5 (also known as Tat-interactive protein-60 (Tip60)), and p300, and smaller complexes with Ikaros family zinc finger protein (IKZF)3, Yin Yang 1 (YY1), and enhancer of zeste homolog 2 (EZH2). These results clearly indicate that FOXP3 orchestrates many cofactors to regulate its targeted genes in order to render T cells with a regulatory function.
2. Association of FOXP3 with transcription factors
Several transcription factors have been identified as FOXP3 cofactors:
Nuclear factor of activated T cells (NFAT): NFAT is important for peripherally induced Tregs (pTregs) or in vitro induced Tregs (iTregs), but not thymic-derived Tregs (tTregs) [15]. NFAT binds to FOXP3 and suppresses NFAT-targeted gene expression [16].
Runt-related transcription factor 1 (RUNX1): RUNX1 interacts with FOXP3 and induces FOXP3 signature genes [17]. In addition, RUNX1 forms a complex with core-binding factor subunit beta (CBFβ) and directly binds to the conserved non-coding sequencing region 2 (CNS2) in the Foxp3 promoter region[18], which is highly de-methylated in Tregs and is important for maintaining FOXP3 expression [18] and for Treg cell lineage stability [19].
NF-κB molecules RelA and c-Rel: c-Rel knockout (KO) mice have a dramatically reduced Treg population, which occurs at the thymic development stage [20]. Rels are also important for pTreg development and FOXP3-regulated gene expression/suppression [21], [22]. Interestingly, the Foxp3-specific deletion of RelA tends to result in more severe autoimmune symptoms than c-Rel deletion, which suggests that RelA is more important than c-Rel in Treg function [22].
Interferon regulatory factor 4 (IRF4): Treg-specific IRF4 deletion results in T helper 2 (Th2)-selective dysregulation and autoimmunity [23].
Together with the transcription factors listed above, many other transcription factors such as Eos (IKZF4) [24], Helios (IKZF2) [14], [25], retinoic acid receptor-related orphan receptor (ROR)γ [26], [27], RORα [28], hypoxia-inducible factor 1-alpha (HIF1α) [29], signal transducer and activator of transcription (STAT)3 [30], GATA3 [13], Kruppel’s associated box-associated protein 1 (KAP1) [31], [32], YY1 [13], [33], and EZH2 [34] have also been identified as FOXP3 cofactors. Among these, HIF1α and YY1 seem to suppress FOXP3 function, while the others induce FOXP3 stability and/or assist FOXP3-targeted gene regulation in Tregs. It is notable that genetic deletion of these genes in Tregs using Foxp3YFP-Cre systems, which was developed by Rubtsov et al. [35], showed some kind of autoimmune phenotypes, but the result was not as severe as in scurfy mice (Table 1) [13], [15], [19], [22], [23], [25], [27], [29], [30], [32], [34]. These facts suggest that the association of those transcription factors with FOXP3 is important, but not critical, for overall FOXP3 function.
Table 1. Phenotypes of genetic deletion of transcription factors in mice.
| Protein name | Mouse model | Phenotype | Ref. |
|---|---|---|---|
| NFAT | Cd4Cre/(NFAT1−/−NFAT4−/−)/NFAT2fl/fl | Impaired iTreg induction; functionally no difference | [15] |
| RUNX-CBFβ | Foxp3YFP-Cre/CBFβfl/fl | Lymphadenopathy and splenomegaly at 5–8 weeks old; not lethal, no obvious sign of autoimmunity up to 8–10 months of age | [19] |
| c-Rel, RelA | Cd4Cre/c-Relfl/fl | Impaired Treg maturation in thymus | [22] |
| Cd4Cre/RelAfl/fl | Slight reduction in Treg population in thymus | ||
| Cd4Cre/c-Relfl/fl/RelAfl/fl | Dramatic reduction in Treg population in lymphoid tissues | ||
| Foxp3YFP-Cre/c-Relfl/fl | No obvious symptoms; impaired Treg function in colitis model | ||
| Foxp3YFP-Cre/RelAfl/fl | Lethal by 15 weeks of age; no population change but impaired function of Tregs | ||
| Foxp3YFP-Cre/c-Relfl/fl/RelAfl/fl | Lethal by 4 weeks of age; dramatic reduction in Treg population and function | ||
| IRF4 | Foxp3YFP-Cre/IRF4fl/fl | Showing autoimmune phenotypes at 3–4 months of age | [23] |
| Helios | Foxp3YFP-Cre/Heliosfl/fl | Not lethal; lymphadenopathy and splenomegaly at 6 months of age | [25] |
| RORγt | Foxp3YFP-Cree/RORCfl/fl | Ameliorate glomerulonephritis in mouse model | [27] |
| HIF1α | Cd4Cre/HIF1αfl/fl | Increased Treg and reduced Th17 population, resistant to EAE model | [29] |
| STAT3 | Foxp3YFP-Cre/STAT3fl/fl | Inflammatory bowel disease symptoms by 12–14 weeks of age | [30] |
| GATA3 | Foxp3YFP-Cre/GATA3fl/fl | Intestinal pathology and dermatitis develop after 6 months | [13] |
| KAP1 | Foxp3YFP-Cre/KAP1fl/fl | Lymphadenopathy and lung inflammation develop at 8–12 weeks of age | [32] |
| EZH2 | Foxp3YFP-Cre/EZH2fl/fl | 50% of lethality by 175 d after birth; showing various autoimmune symptoms | [34] |
EAE: experimental autoimmune encephalomyelitis; −/−: double KO; fl/fl: flox/flox, floxed; YFP: yellow fluorescent protein; Cre: Cre recombinase.
3. Association of FOXP3 with post-translational modulators
FOXP3 is regulated by post-translational modifications (PTMs) [36], which include phosphorylation, acetylation, ubiquitination, and methylation, among others. Compared with transcription factors discussed above, conditional deletion of PTM proteins in Tregs tends to result in more severe autoimmune phenotypes (Table 2) [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49]. However, these PTM proteins also regulate cell functions by modifying histone and other proteins epigenetically. It remains to be elucidated whether PTM of FOXP3 alone is sufficient for Treg stability and function, or whether epigenetic modifications on any other proteins also contribute.
Table 2. Phenotypes of genetic deletion of post-translational modifications in mice.
| Protein name | Mouse model | Phenotype | Ref. |
|---|---|---|---|
| Tip60 | Foxp3YFP-Cre/Tip60fl/fl | Lethal by 4 weeks of age; significant reduction in Treg population and function | [37] |
| CBP/p300 | Foxp3YFP-Cre/EP300fl/fl | Mild autoimmunity develops at 10 weeks of age; less Treg-suppressive function | [38] |
| Foxp3YFP-Cre/CREBBPfl/fl | 50% of mice have enlarged peripheral lymphoid tissues at 8–10 weeks of age; less Treg function | [39] | |
| Foxp3YFP-Cre/ (CREBBPfl/flEP300fl/fl) | Lethal by 4 weeks of age; dramatic reduction in Treg population and function | [39] | |
| HDAC3 | FOXP3YFP-Cre/Hdac3fl/fl | Lethal by 6 weeks of age; impaired Treg function | [40] |
| HDAC6 | KO | Increased Treg-suppressive function; regulated FOXP3 acetylation | [41] |
| HDAC9 | KO | Increased Treg-suppressive function | [42] |
| HDAC11 | Foxp3YFP-Cre/Hdac11fl/fl | Increased Treg-suppressive function; regulated FOXP3 acetylation | [43] |
| SIRT1 | Foxp3YFP-Cre, Cd4Cre/Sirt1fl/fl | Increased Treg-suppressive function; regulated FOXP3 acetylation | [44] |
| Pim2 | KO | Increased FOXP3 expression and Treg-suppressive function | [45] |
| CDK | KO | Increased Treg-suppressive function | [46] |
| Cbl-b | KO | Increased Treg population | [47] |
| USP7 | Foxp3YFP-Cre/USP7fl/fl | Lethal by 4 weeks of age; significant reduction in Treg population and function | [48] |
| USP21 | Foxp3YFP-Cre/USP21fl/fl | Lymphadenopathy and splenomegaly develop at the age of 6–8 months | [49] |
| PRMT5 | Foxp3YFP-Cre/PRMT5fl/fl | Lethal by 4 weeks of age; significant reduction in splenic Treg and overall Treg function | —a |
CBP: cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB)-binding protein; HDAC: histone deacetylase; SIRT1: sirtuin-1; CDK: cyclin-dependent kinase; Cbl-b: Casitas-B-lineage lymphoma protein-b; USP: ubiquitin-specific peptidase; PRMT: protein arginine methyltransferase.
- a
-
Unpublished data by Nagai et al.
3.1. Acetylation and deacetylation of FOXP3
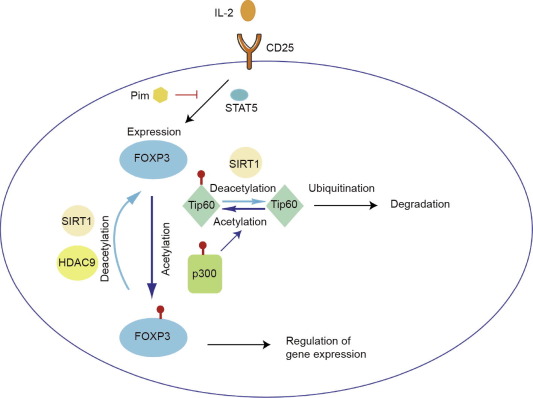
FOXP3 is known to be acetylated by KATs. KATs can add acetyl groups to the lysine of their target proteins. Among KATs, Tip60 (KAT5) and cyclic adenosine monophosphate response element-binding protein (CREB)-binding protein (CBP)/p300 (KAT3A/3B) directly bind to FOXP3 and acetylate it, which enhances DNA binding and stability [38], [50], [51].
Since acetylation sites in FOXP3 overlap with ubiquitination sites, acetylation by KATs prevents ubiquitin-dependent proteasomal degradation (as described below) and induces the stability of FOXP3 protein.
We have found that Tip60 and p300 acetylate each other and further induce FOXP3 acetylation [37]. A structure model suggests that the single acetylation of Tip60 at K327 by p300 would favor FOXP3 binding to the Tip60 cleft. Genetic deletion of Tip60 in Tregs demonstrated severe autoimmune problems similar to those of scurfy mice, suggesting that Tip60 is essential for Treg function.
We have also reported that the natural p300 inhibitor garcinol induces FOXP3 degradation via a lysosome-dependent pathway, which suggests that p300 plays a role in lysosome-dependent FOXP3 degradation [52]. However, single deletion of p300 [38] or CBP [39] only causes relatively mild autoimmunity. Since CBP and p300 have high homology and are functionally conserved [53], single deletion is not effective to limit p300 or CBP function. This has been confirmed by double deletion of p300 and CBP, which causes severe autoimmunity on a level similar to the deletion of Tip60 [39].
Lysine deacetylase, which is sometimes known as histone deacetylase (HDAC), also regulates FOXP3 functions. HDAC consists of four different classes based on the cellular location, size, number of catalytic pockets, and homology to yeast prototypes [54]:
-
•
Class I: HDAC1, 2, 3, and 8;
-
•
Class II: HDAC4, 5, 6, 7, 9, and 10;
-
•
Class III: SIRT1–7;
-
•
Class IV: HDAC11.
Class I HDACs appear to enhance Treg functions. Conditional deletion of HDAC3 in Tregs results in decreased Treg function and severe scurfy-like phenotypes [40]. Other Class I HDACs, namely HDAC1 and HDAC2, also bind FOXP3 at the N terminal to regulate the expression of many genes [55]. These Class I HDACs do not seem to alter FOXP3 PTM; rather, they seem to be recruited by FOXP3, which in turn counteracts the hyper-acetylation of the promoter, and thereby switches off inflammatory genes.
Li et al. [55] have found that FOXP3 binds directly to Tip60, HDAC7, and HDAC9 at the N-terminal region to form a chromatin-remodeling complex. Although the association of HDAC7 with FOXP3 and Tip60 seems to be important in order for FOXP3 to repress the targeted gene [56], the precise mechanism is as yet unknown. Since HDAC7 has little enzymatic activity and needs to form a complex with other factors such as HDAC3 in order to function [40], HDAC7 may be important in recruiting other FOXP3 cofactors. However, deletion of HDAC6 [41], [57], HDAC9 [42], [57], sirtuin-1 (SIRT1) [44], [57], [58], [59], or HDAC11 [43] increases the acetylation level and overall expression of FOXP3, in addition to enhancing Treg functions.
3.2. Interaction of FOXP3 with kinases
Phosphorylation of FOXP3 was first described by Samanta et al. [60]. Since then, several kinases, such as cyclin-dependent kinase (CDK)2, Pim1, Pim2, and lymphocyte-specific protein tyrosine kinase (LCK), have been reported to phosphorylate FOXP3 [18], [36]. Phosphorylation of FOXP3 by CDK2 [46], [61], Pim1 [62], and Pim2 [45] decreases FOXP3 function. CDK-deficient Tregs show higher suppressive functions than wild-type cells. Mutation of the CDK binding site on FOXP3 increases FOXP3 expression and function.
Pim1 phosphorylates FOXP3 at S418, which reduces the chromatin binding of FOXP3 to targeted genes, and prevents further phosphorylation at S422 of FOXP3, which is reported to be important for FOXP3 function [63]. Pim2 is primarily identified as a FOXP3-dependent gene and is involved in Treg expansion in vitro [64]. We have found that Pim2 interacts with FOXP3 directly, and phosphorylates it at the N-terminal site [45]. Pim2 KO mice have Tregs with higher FOXP3 expression and enhanced suppressive function, and are resistant to dextran sodium sulfate (DSS)-induced colitis; this suggests that phosphorylation by Pim2 also affects FOXP3 stability and function. LCK phosphorylates FOXP3 at Y342 in cancer cells and upregulates its function [65]. However, the mechanism by which LCK interacts with FOXP3 and affects subsequent phosphorylation is as yet unknown.
Phosphorylation of FOXP3 at S418 by an unknown kinase augments the DNA binding of FOXP3 to chromatin [63]. Dephosphorylation of this site by tumor necrosis factor (TNF)-induced protein phosphatase I (PP1) has been observed in arthritis patients, whose Tregs show markedly impaired suppressive function [63].
3.3. Ubiquitination and degradation of FOXP3
Protein ubiquitination is a multistep process that is regulated by the E1, E2, and E3 ligase families. Ubiquitination is caused by the binding of ubiquitin peptides on the lysine residue of the targeted protein. On its own, the ubiquitin peptide has seven lysine residues, including K48 and K63. At least one of these lysine residues on each ubiquitin peptide must be utilized in order to form a polyubiquitin chain [66]. Lysine-48-linked (K48) polyubiquitination is the dominant form that marks proteins for degradation by the 26S proteasome, while non-K48 polyubiquitinations are usually implicated in regulating the targeted protein’s function, rather than in protein degradation [67]. The majority of FOXP3 ubiquitination is related to K48 ubiquitination and degradation [18], [36].
Several pathways induce FOXP3 ubiquitination. HIF1α, which is induced under hypoxic conditions and directly binds to FOXP3, induces FOXP3 polyubiquitination and degradation [29]. Chemokine (C–C motif) ligand 3 (CCL3) signaling also induces polyubiquitination by activating protein kinase B alpha (PKBα) pathways in the psoriatic microenvironment; however, the precise mechanism is still unclear [68]. During T cell receptor (TCR) stimulation, selective inhibition of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-AKT signaling pathway was found to suppress the proliferation of Tregs, but not of conventional T cells to any significant degree; this suggests that Tregs are more dependent on PI3K-AKT pathways, and that AKT inhibitors can be used to limit Treg infiltration into tumors and to induce immunity against tumors, if they are able to be delivered specifically to Treg [69].
The enzymes that ubiquitinate FOXP3 for degradation are the C terminus of HSC70-Interacting Protein (CHIP) and Casitas-B-lineage lymphoma protein-b (Cbl-b). Chen et al. [70] have shown that STUB1 causes K48 type ubiquitination of FOXP3 at residues K227, K250, K263, and K268 in a heat shock protein (HSP)70-dependent manner. Specific knockdown of STUB1 or HSP70 increases FOXP3 expression, and STUB1 overexpression in Tregs decreases FOXP3 expression level and results in reduced suppressive Treg function in both in vitro studies and in vivo mouse models. Cbl-b also binds to FOXP3 upon TCR stimulation, and, in collaboration with STUB1, ubiquitinates and subsequently degrades FOXP3 [47].
As polyubiquitination reduces FOXP3 stability, it is expected that deubiquitinases will increase FOXP3 stability. Ubiquitin-specific peptidase(USP)7 [48], [71] and USP21 [49] have been reported to directly interact with FOXP3 to induce stability by degrading ubiquitin. In a demonstration of the importance of USP7 and USP21, Treg-specific deletion of either of these peptidases was shown to cause autoimmune reaction in mice [48], [49]. It is interesting to note that although both of these deubiquitinases cause autoimmunity, the deletion of USP7 resulted in a more severe phenotype. In those Tregs, Tip60 expression was also dramatically reduced, indicating that this severity is also partially due to Tip60 malfunction.
3.4. Methylation of FOXP3
A recent mass spectrometric approach has shown that FOXP3 can be di-methylated. Using isomethionine and specific arginine methylation antibody, Geoghegan et al. [72] identified 2502 arginine methylation sites from 1257 tissue-specific and housekeeping proteins in human T cells. From these peptides, they identified a FOXP3 peptide that is methylated at the R51 position. Working separately, we identified protein arginine methyltransferase (PRMT)5, which symmetrically di-methylates arginines [73], as a novel cofactor of FOXP3†. Conditional deletion of PRMT5 in Tregs resulted in a severe scurfy-like phenotype, suggesting that FOXP3 methylation is also critical for FOXP3 functions.
4. Therapeutic applications targeting FOXP3 and its cofactors
Because Tregs are the dominant T cells that suppress host immunity, they are considered to be an important target for clinical applications in autoimmunity [74] and cancer immunotherapy [75]. For this purpose, many inhibitors and modulators have been tested in attempts to directly regulate FOXP3 cofactors or the expression of the FOXP3 protein (Fig. 1). However, most of these studies have been performed in animal models, as proofs of concept. Only a few have been tested in clinical settings.
 Fig. 1. A roadmap for targets to regulate FOXP3 and Treg functions. IL: interleukin.
Fig. 1. A roadmap for targets to regulate FOXP3 and Treg functions. IL: interleukin.