1. Introduction
Antimony (Sb) has a long and diverse history of applications, from a marginal use in cosmetics, pigments and pharmacology in ancient times to a drastic increase in its demand over the 20th century, especially for the fabrication of military artefacts and munitions during war periods, and as the main compound in lead-acid batteries for mass production in the automotive industry. More recently, antimony has been increasingly employed in the fabrication of flame retardants (heavy textiles, plastics, rubber, paints, etc.) and also as a catalyst for polyethylene terephthalate (PET) production and as a lead alloy component, among several other applications (Anderson, 2019, Anderson, 2001, European Commission, 2017a, Schwarz-Schampera, 2014).
As environmental and supply risk concerns have increased compared to much earlier decades, there has also been a growing interest in the supply of antimony from secondary sources, including used lead-acid batteries (Anderson, 2019, Anderson, 2001; Schwarz-Schampera, 2014; USGS, 2020a) and residues generated during the processing and refining of economically valuable metals such as copper, lead and gold (Anderson, 2019, Anderson, 2001, Schwarz-Schampera, 2014).
A number of factors, however, might affect the supply of this valuable resource for future generations. These include geopolitical, economic, social, environmental, and technological aspects, such as:
-
•
China continues to be the major supplier of antimony worldwide, and current production is still limited to a few countries (Anderson, 2019, European Commission, 2017b, European Commission, 2014, European Commission, 2010, USGS, 2020a). More than 60–70% of the antimony production is supplied by China, although there are some other countries (e.g. Bolivia, Russia, Australia, Serbia, Slovakia, Bulgaria, Greenland, Turkey, Thailand, Kyrgyzstan, South Africa, Tajikistan) which might be relevant exporters of antimony (e.g. ore concentrate, refined metal) in the future, based on their estimated reserves (Anderson, 2019, European Commission, 2017a, Grund et al., 2011, Lauri, 2018, Schwarz-Schampera, 2014; USGS, 2020a, USGS, 2020b);
-
•
The recycling rates of this metal are still low (below 30%) and mainly restricted to used lead-acid batteries (European Commission, 2017a, Grund et al., 2011, Sundqvist Ökvist et al., 2018, USGS, 2020a, USGS, 2020b), whose availability could decrease over time due to the use of alternative technologies;
-
•
The substitution of antimony trioxide for its main application in flame retardants might be challenging when high performance is required (European Commission, 2017a, European Commission, 2010, Schwarz-Schampera, 2014), although some estimates suggest that this measure could be feasible in the medium term (Henckens et al., 2016); and
-
•
Antimony is becoming more relevant for emerging applications including micro-capacitors (European Commission, 2010, Sinding-Larsen and Wellmer, 2012), the semiconductor industry (Multani et al., 2016; Schwarz-Schampera, 2014), emerging thin film photovoltaic cells (Kondrotas et al., 2018, Nair et al., 2018, Yang et al., 2019), and as an anode material for sodium-ion storage batteries (Li et al., 2019); this may give a potential rise to its demand in the future.
The vulnerability of Europe, in particular, to the antimony supply risks has influenced the European Commission to seriously consider this matter. In that sense, the European Commission has carried out a series of assessments (European Commission, 2020, European Commission, 2017b, European Commission, 2014, European Commission, 2010) where a selected group of raw materials has been considered as “critical”, and antimony has persistently been positioned on the criticality list so far.
Antimony is considered to be found in the Earth’s crust at an abundance of approximately 0.2 ppm (Anderson, 2001; Schwarz-Schampera, 2014), requiring an enrichment of about 150000 times its crustal content to achieve a concentration that can be considered as potentially economic (about 3% antimony by weight) (Schwarz-Schampera, 2014). Although this metalloid has been found in more than two hundred antimony-bearing mineral phases (Multani et al., 2016; Schwarz-Schampera, 2014), its sourcing from mining activities has been largely from stibnite or antimonite (Sb2S3) (Anderson, 2019, Anderson, 2001, European Commission, 2017a, Lager and Forssberg, 1989a, Morizot and Ollivier, 1993, Richards, 1977, Schwarz-Schampera, 2014). Some of the major stibnite-bearing deposits known are the Xikuangshan deposit in Hunan, China, and the Antimony Line, Murchison Belt in South Africa (Schwarz-Schampera, 2014).
Stibnite frequently occurs in greenstone-hosted deposits comprising quartz-carbonate veins and carbonate replacements (1.5–25% Sb2S3; lacking significant base or precious metals). It also occurs, in minor proportion, in epithermal gold-antimony veins (0.1–3.5% Sb2S3; low-sulfidation subtype enriched in As, Hg, Au, Ag, Te), reduced-magmatic systems (approximately 0.1–1.5% Sb2S3; associated to metals such as Au, Bi, Te, W, As, Ag, Pb, Zn), polymetallic base metal veins (0.1–0.5% Sb2S3; enriched in Ag), and hot springs (0.1–0.2% Sb2S3; co-enrichment with As and Hg; might occur along with Te, Se, Au, Ag) (European Commission, 2017a; Schwarz-Schampera, 2014). When stibnite has been exposed to the surface it is usually weathered to oxides including bindheimite, kermesite, stibiconite and valentinite. For a comprehensive description of antimony-bearing deposits, the reader is referred to Schwarz-Schampera (2014).
The exploitation and processing of stibnite-bearing ores involve the use of conventional stages of mining (open-pit or underground), comminution (crushing and grinding) and concentration techniques including gravity separation and froth flotation (Anderson, 2019, Anderson, 2012, European Commission, 2017a, Schwarz-Schampera, 2014). This allows an antimony-enriched concentrate to be obtained, the recovery of which will depend on the efficiency along the mining and processing chain. Considerable research efforts have been devoted in recent years to cover mineralogical and mineral processing aspects of antimony ores, with a greater emphasis on hydro- and pyrometallurgical processing of antimony concentrates (Anderson, 2019, Grund et al., 2011, Rohner and Millard, 2016, Schwarz-Schampera, 2014; Solozhenkin and Alekseev, 2010a, Yang et al., 2018), and to a lesser extent on stibnite froth flotation.
While relevant yet outdated reviews on stibnite froth flotation were carried out in the 1970s and 1980s, there has been no attempt in the literature to provide a critical review on progress over the last three decades. Richards' pioneering work in the 1970s (Richards, 1977), for example, combined a general overview of previous literature (i.e. patents, journal articles, reports, etc.) related to stibnite froth flotation with experimental work at laboratory-scale to establish optimal conditions for obtaining a stibnite concentrate from an ore deposit in New Zealand. Just over a decade later, Lager and Forssberg, 1989a, Lager and Forssberg, 1989b carried out an extensive analysis of published studies (including Richards' work (Richards, 1977)) concerning the flotation of stibnite and other antimony minerals. The authors summarised relevant findings regarding the effect of key process variables (e.g. pulp pH, collector type) and approached a selected number of case studies in mine locations around the world.
Other work for specific locations includes, for instance, a study of physicochemical and technical aspects related to the flotation of complex antimony ore deposits in Russia (Solozhenkin and Alekseev, 2010b, Solozhenkin and Kovalev, 2018); a discussion on flotation techniques employed at the Consolidated Murchison mine in South Africa (Grund et al., 2011, Davis et al., 1986); and the description of technological aspects of froth flotation for a stibnite ore from Xikuangshan and some other locations in China (Hu, 1988, Anderson, 2012).
On the basis of the aforementioned considerations and to the best of the authors' knowledge, a comprehensive and updated assessment focusing on key aspects of the froth flotation of stibnite ores is still lacking in the literature. This is particularly relevant considering the increasing concerns in the supply of antimony from primary sources in the medium- and long-term. The present work aims to fill this gap by exhaustively revising the literature regarding the technological and environmental aspects related to froth flotation of stibnite-bearing ores. These include: surface chemistry and flotation reagents (Section 2), particle and mineralogical effects (Section 3), flowsheet design (Section 4), and topics related to antimony speciation in the environment, as well as a broader utilisation of antimony resources via froth flotation (Section 5). Finally, some concluding remarks are highlighted in Section 6. Through this approach, the authors aim to contribute to the dissemination of best practices towards a more sustainable supply of antimony (ore concentrate) from primary, and potentially secondary sources.
2. Surface chemistry and reagents in stibnite froth flotation
The first part of this section (Subsection 2.1) is concerned with a brief review of selected studies that have aimed to deepen the fundamental understanding of the interactions between the aqueous medium (i.e. water and flotation reagents/bioreagents) and the stibnite surface and how these affect the floatability of stibnite and its selectivity with respect to certain gangue minerals. Such studies have been carried out at the microscale, usually relying on a few grams of medium-to-high-purity, carefully prepared ore sample. In subsequent sections (2.2 Frother effect, 2.3 Collector effect, 2.5 pH regulator effect, 2.6 Depressant effect, 3 Mineralogy and particle size effects in stibnite froth flotation, 4 Flowsheet design for stibnite recovery by froth flotation) the review is mainly focused on discussing the research progress in stibnite froth flotation at both bench- and industrial-scale.
2.1. Floatability, electrokinetic behaviour and surface properties of stibnite at the microscale
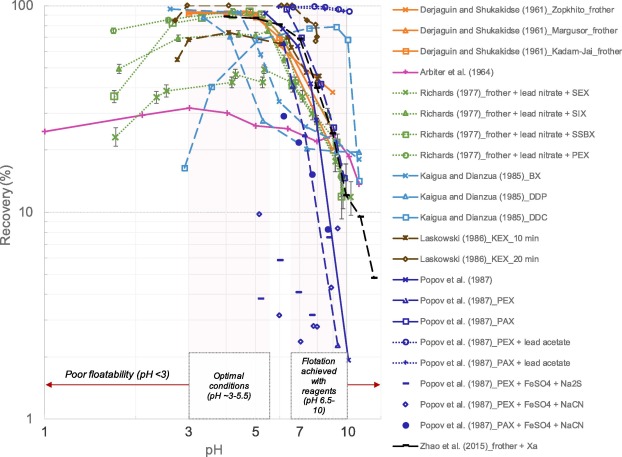
According to Kelebek (1984), the study carried out by Derjaguin and Shukakidse (1961) was the first to report the remarkable effect of pH on the floatability and electrokinetic (zeta) potential of stibnite. A few years later, Arbiter et al. (1964) extended the scope of Derjaguin and Shukakidse’s work to other “hydrophobic” minerals in addition to stibnite and complemented the microflotation and electrokinetic measurements with the estimation of the contact angle at various pH values. Unlike the previous study, Arbiter et al. (1964) did not use a frother in the microflotation tests, which certainly led to a much lower stibnite recovery over the entire pH range (Figure 1); however, the purity of their samples was not reported. On the other hand, Popov et al. (1987) showed a relatively good natural floatability of nearly pure stibnite at a pH less than 6.5 (Figure 1).
 Fig. 1
Fig. 1Several other authors have also studied the floatability behaviour of stibnite as a function of pH at the microscale by introducing a series of variations in their tests: ore type (Derjaguin and Shukakidse, 1961), addition of reagents (frother, collector, activator, depressant) (Kaigua and Dianzua, 1985, Popov et al., 1987, Richards, 1977), or flotation time (Laskowski, 1986). What has become evident from all these results (Figure 1) is that:
-
•
Extremely acidic or alkaline pH ranges (<3 or >10, respectively) are not favourable for the recovery of stibnite;
-
•
An acidic pH (∼3–5.5) seems the most favourable for the floatability of stibnite, especially with conventional reagents (e.g. xanthate collector, lead nitrate as an activator);
-
•
It is possible to extend the floatability range of stibnite at slightly alkaline values (<10) with the use of certain reagents (lead acetate as an activator, diethyl dithiocarbamate as collector);
-
•
It is also possible to inhibit the floatability of stibnite in a slightly acidic to slightly alkaline pH range (∼6–9) with the use of some depressants (FeSO4 in combination with Na2S or NaCN).
On the other hand, Arbiter et al. (1964) explained the zeta potential and contact angle behaviour of stibnite at different pH values on the basis of the crystal structure of stibnite. It was, in fact, presumed that the existence of weak Sb-S bonds may lead to the development of ionic sites in the stibnite surface at alkaline pH (formation of SbOH and SH sites in equilibrium with SbO− and S− sites depending on the pH). The reversible proportion of these ionic sites could have explained the decrease in the contact angle of stibnite as pH was increased (Arbiter et al., 1964), as observed in Figure 2a. Overall, most of the contact angle measurements reported by different authors showed a similar trend; that is, contact angle remains relatively stable within a range between 30° and 44° at acidic pH (1< pH <7), while it tends to decrease as pH turns alkaline (Figure 2a). Moreover, the addition of collector and activator sharply increased the contact angle of stibnite (up to approximately 86°) and enhanced its floatability at a pH range from slightly acidic to alkaline (6< pH <11), on the basis of the work conducted by Vijayakumar and Majumdar (1972) apud Lager and Forssberg (1989a) (Figure 2a).
 Fig. 2
Fig. 2Nevertheless, there was some variability in the data reported (Figure 2a) that can be explained by the variation in materials and experimental procedures. In fact, the adoption of different methods for sample preparation clearly influenced the contact angle measurements in the results reported by Kelebek (1984), as observed in Figure 2a. Starting from the point that there is no contamination in the liquid or on the solid surface, physical and chemical aspects of the surface such as surface roughness (Arbiter et al., 1964, Chau et al., 2009, Ozcan, 1992), surface heterogeneity and surface geometry (Chau et al., 2009, Ozcan, 1992) have been highlighted as determining factors in the estimation of contact angles. Zeta potential measurements of stibnite (Figure 2b) have, on the other hand, indicated that its surface is negatively charged within a wide pH range, allowing positive ions in solution to be attracted to the surface (Ealedona and Fujita, 2006).
A detailed analysis of the Pourbaix diagram allows gathering insights into the predominance of antimony compounds on aqueous media as a function of potential (Eh) and pH. As observed in Figure 3, the ionic species SbO3−(aq) predominates over a wide Eh and pH range, particularly in more oxidising conditions. On the contrary, stibnite is more stable at reducing conditions and in an acidic to slightly alkaline pH. It is also clear that, at an alkaline (pH > 8) and reducing environment, stibnite dissolves and is transformed into the ionic species Sb2S42−(aq) (Vink, 1996). These observations indicate stibnite amenability to float more effectively at acidic pH, with its floatability diminishing at alkaline pH, as will be discussed in Section 2.5. This diagram (Figure 3) is, however, not considering the effect of flotation reagent addition.
 Fig. 3
Fig. 3While the Pourbaix diagram will vary depending on the thermodynamic data available, the assumptions made regarding the activity of the species, and the number of species considered, it provides useful information on the occurrence of species at either oxidising or reducing environments over the entire pH range. Notably, to the best of the authors’ knowledge, there are no reported studies in the literature on the use of Pourbaix diagrams to support the interpretation of Eh and pH measurements for stibnite flotation. However, Eh-pH diagrams have been published for a wide range of species that typically are of interest in the context of stibnite flotation (e.g. the As-O-H-S-Fe system, as shown by Lu and Zhu, 2011, Vink, 1996).
Cao et al. (2018a) evaluated the effect of lead nitrate addition on the conditioning of stibnite at a pH of 6.5 and found that more than 89% of the lead added was adsorbed on the stibnite surface, mainly as Pb2+ ions. Interestingly, their results showed an intermediate dosage of activator for which the adsorption of lead was optimal (99.8% of Pb adsorbed on stibnite for an activator dosage of 5 × 10−5 mol/L). Additionally, they reported a slight increase of Sb in solution (up to 3.42 × 10−5 mol/L of Sb) as the activator dosage was increased, in relation to the initial concentration of Sb in deionised water (1.77 × 10−5 mol/L of Sb). This was believed to occur, according to the authors, due to the substitution of Sb ions by Pb ions on the stibnite surface. Furthermore, Cao et al. (2018b) showed that both copper sulphate and lead nitrate can modify the zeta potential of stibnite in deionized water at a pH of 6.5 (from −22.1 mV to nearly 0 mV), as the concentration of any of these reagents is increased (from 0 to 1 × 10−4 mol/L). The adsorption of Pb2+ or Cu2+on the negatively charged stibnite surface was attributed as the reason for the increase in zeta potential of stibnite. The authors, however, did not carry out zeta potential measurements at higher activator dosages. Such a study would have been interesting in the context of stibnite stability (Figure 3) with metal ions (Pb2+ or Cu2+) in solution in a less reducing, activator-induced environment. Furthermore, the effect of the collector-activator system on stibnite flotation could also be examined and supported by zeta potential measurements and Pourbaix diagram interpretation.
Some microflotation studies have also aimed to understand the effect of the reagent dosage on the recovery of stibnite. It has been shown that lead nitrate has a somewhat superior performance compared to copper sulphate in the activation of the stibnite surface, according to the comparative tests carried out by Richards, 1977, Cao et al., 2018b at acidic pH (5.0 and 6.5, respectively). On the other hand, in the study by Qing and Ai (1988), copper sulphate achieved a good activation performance (slightly higher recovery of stibnite than the one obtained with lead nitrate in the aforementioned studies) in the flotation of a stibnite-bearing ore containing arsenopyrite at natural pH (no pH regulator added) and in combination with an organic depressant. The results of those tests have been summarised in Figure 4a.
 Fig. 4
Fig. 4The role of collector dosage on the microflotation of stibnite has also received some attention (Cao et al., 2018a, Richards, 1977). It has been demonstrated that short-chain xanthates such as sodium ethyl xanthate require a much higher concentration to achieve the performance obtained with long-chain xanthates at acidic pH. The aforementioned studies also confirmed that the activation of the mineral surface is essential for the adequate recovery of stibnite, at least when xanthate collectors are employed (Figure 4b).
A few researchers have also attempted to elucidate the possible mechanisms responsible for the alteration of the surface properties of stibnite when it comes into contact with reagents. For this purpose, a series of qualitative and quantitative techniques have been used to characterise the products formed at the liquid–solid interface, as well as the compounds in the solution (Table 1). Popov et al. (1987) analysed the infrared spectra of stibnite surface products and concluded that potassium amyl xanthate alone slightly improves the floatability of stibnite at acidic pH due to the formation of antimony xanthate on the mineral surface. On the other hand, they found that potassium ethyl xanthate (PEX) alone did not improve the floatability of this mineral as it was not adsorbed without activator. In the latter case, stibnite recovery in the concentrate was more likely attributed to the natural floatability of the nearly pure stibnite sample (Table 1), rather than to the addition of collector (i.e. PEX). Moreover, the addition of lead acetate allowed the formation of lead xanthate on the stibnite surface, which is responsible for the flotation of stibnite even at alkaline pH (Figure 1). Richards (1977) attributed the chain-length effect of xanthate collector on stibnite flotation performance to increased adsorption of the collector species on the mineral (at the mineral surface-liquid interface), due to lateral interaction between the adsorbed species. Richards also highlighted that the increase in carbon chain length of xanthates may affect the selectivity and, consequently, the antimony grade of the final concentrate in more complex stibnite-bearing ores, although this was not tested in the experiments reported by the author.
Table 1. Summary of the research techniques used in studies related to the characterisation of the floatability, electrokinetic behaviour and surface properties of stibnite at the microscale, with an emphasis on microflotation tests.
| Source | Reported research methods 1 | Microflotation test conditions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell type/ volume (mL) 2 | Ore sample | Reagents | Max. recovery, % 5 | ||||||||
| Mass, g | Size range, µm | Purity,% | Frother | Collector 3 | Activator | pH Regulator | Depressant | ||||
| Derjaguin and Shukakidse (1961) | MF; EP | (70) | – | 150 – 43 | – | n-hexyl alcohol | – | – | H2SO4; NaOH | – | 92 – 93 (pH 3, 2 min) |
| Arbiter et al. (1964) | MF; CA | B | – | 150 – 44 | – | – | – | – | – | – | 31.8 (pH 3, 8 min) |
| Vijayakumar and Majumdar (1972) apud Lager and Forssberg (1989a) | CA | – | – | – | – | – | PIAX | Pb(NO3)2 | – | – | – |
| Richards (1977) | MF; XRD; MA; AAS; UVS | A (120); D (250) | 25 (cyl.) | 500 – 50; 150 – 63 (Hall.); 147 – 74 (cyl.) | ∼28.5 (Sb) | Aerofroth 65/70/71/73/77; pine oil; cresylic acid | SEX; SIPX; SSBX; PEX; PAX | Pb(NO3)2; CuSO4 | H2SO4, Na2CO3 | – | ∼94.4 (pH 5, PAX, Pb(NO3)2, 5 min) |
| Kelebek (1984) | CA; AASP | – | – | – | – | – | – | – | – | – | – |
| Kaigua and Dianzua (1985) apud Lager and Forssberg (1989b) | MF | A | – | – | – | – | BX; DDP; DDC | – | – | – | ∼96 (pH ∼ 2.6, BX); ∼87 (pH ∼ 3.4, DDP); ∼79 (pH ∼ 9.3, DDC) 6 |
| Laskowski (1986) | MF | B | – | 200 – 75 | – | – | PEX | – | H2SO4; KOH | – | ∼74 (pH ∼ 4, 10 min); ∼100 (pH ∼ 3.0 – 6.6, 20 min) |
| Popov et al. (1987) | MF; IRS | B (100) | 1 | 100 – 75 | 70.27 (Sb) | – | PEX; PAX | Pb(C2H3O2)2 | H2SO4; NaOH | FeSO4 + Na2S; FeSO4 + NaCN | ∼99.9 (pH ∼ 5.2, PAX, Na2S, 5 min) |
| Qing and Ai (1988) | MF | – | 2 | < 75 | 95.2 (Sb2S3) | Pine oil | BX +“SN No. 9” | CuSO4 | – | “Organic depressant B” | ∼90 (natural pH) |
| Leming et al. (1998) | MF; EP; UVS | – | – | – | 35.34; 54.31; 32.89 (Sb) 4 | – | BX | – | NaOH or Na2CO3 | Na2S2O4 (reducing agent) | ∼97 (pH 9.8, AM); ∼88 (pH 9, NBC) 4 |
| Ealedona and Fujita (2006) | EP | – | – | – | – | – | – | – | – | – | – |
| Zhao et al. (2015) | MF; DFT | (50) | 2 | – | 96.5 (Sb2S3) | Pine oil | Xa | – | NaOH | – | ∼88 (pH ∼ 4, 5 min) |
| Matveeva et al. (2017) | MF; LSM; UVS | C (20) | 1 | 100–63 | 48.4 (Sb) | Pine oil | DDC + OPDTC | – | – | Tannin | ∼68–70 |
| Cao et al. (2018a) | MF; ICP-MS; DFT | C (100) | 3 | 150–104 | > 99 (Sb2S3); 71.6 (Sb) | Pine oil | BX | Pb(NO3)2 | HCl; NaOH | – | ∼90 (pH 6.5, 2 min) |
| Cao et al. (2018b) | MF; EP; XPS; ToF-SIMS | C (40) | 2 | 75–63 | Pb(NO3)2; CuSO4 | ∼85.3 (pH 6.5, Pb(NO3)2, 1 min) | |||||
- 1
-
MF = Microflotation; EP = Electrokinetic (zeta) potential; CA = Contact Angle; XRD = X-ray Powder Diffraction; MA = Microscopy Analysis; LSM = Laser Scanning Microscopy; AAS = Atomic Absorption Spectrometry; ICP-MS = Inductively Coupled Plasma Mass Spectrometry; ToF-SIMS = Time-of-Flight Secondary Ion Mass Spectrometry; AASP = Atomic Absorption Spectrophotometry; UVS = Ultraviolet–visible Spectrophotometry; IRS = Infrared Spectrophotometry; XPS = X-ray Photoelectron Spectroscopy; DFT = First-principles calculations based on Density Functional Theory.
- 2
-
A = Hallimond cell; B = Modified Hallimond cell; C = Mechanical cell; D = Measuring cylinder, used by Richards (1977) in frother tests.
- 3
-
PIAX = Potassium isoamyl xanthate; SEX = Sodium ethyl xanthate; SIPX = Sodium isopropyl xanthate; SSBX = Sodium sec-butyl xanthate; PEX = Potassium ethyl xanthate; PAX = Potassium amyl xanthate; BX = Sodium or potassium butyl xanthate; Xa = Unspecified ethyl xanthate; DDP = Dibutyl dithiophosphate; DDC = Diethyl dithiocarbamate; OPDTC = Oxypropyl diethyldithiocarbamate ester.
- 4
-
MF tests carried out on artificial mixtures (AM) of high-purity minerals (stibnite and arsenopyrite) and on a natural stibnite-arsenopyrite bulk concentrate sample (NBC).
- 5
-
The concentrate grade is not consistently reported in most of the studies. Flotation time corresponding to the maximum recovery of stibnite (or antimony) has also been included when available.
- 6
-
Low selectivity of stibnite in relation to arsenopyrite when using BX or DDP and no activation. The best selectivity was achieved with DDC (<15% arsenopyrite in the concentrate).
Leming et al. (1998), on the other hand, combined microflotation and electrokinetic potential measurements with UV spectrophotometry. Pulp potential measurements on pure mineral samples at pH 9.2 and with the addition of butyl xanthate (5 × 10−4 mol/L) revealed that arsenopyrite floats in the range −200 to 200 mV, while stibnite is heavily depressed below 0 mV due to the reduction of butyl dixanthogen on its surface. Accordingly, the authors concluded that stibnite and arsenopyrite could be, in theory, separated at alkaline pH and in the potential region of −200 to 0 mV. Further microflotation tests carried out with artificial mixtures of both minerals and with a natural stibnite-arsenopyrite bulk concentrate sample confirmed that stibnite and arsenopyrite can be effectively separated at pH 9–10. The separation was achieved by controlling the pulp potential within a negative range (from −150 to −120 mV) and using Na2S2O4 as the reducing agent of the hydrophobic compound (butyl dixanthogen) on the stibnite surface. Key experimental details regarding the electrode system (e.g. reference electrode type) were, however, not reported in their study.
More recent studies (Cao et al., 2018a, Zhao et al., 2015) have attempted to explain the surface properties of stibnite on the basis of its crystal structure and with the aid of first-principles calculations supported by Density Functional Theory (DFT). Zhao et al. (2015) postulated that the floatability differences between stibnite and jamesonite at high pH may be related to the coordination structures of Sb atoms and that the presence of metal atoms in the Highest Occupied Molecular Orbital (HOMO) favours the depression of both minerals with lime. Cao et al. (2018a) established that Pb2+ is the dominant species that is adsorbed on five possible sites on the stibnite surface and provides a stronger interaction of the xanthate with the activated stibnite surface, in contrast to the adsorption of butyl xanthate at Sb sites on a non-activated surface.
2.2. Frother effect
The most frequently used frothers in stibnite froth flotation according to literature are pine oil (natural oil-type frother, also referred to as 2# oil by Wang (2016)) and a polyglycol-type frother commercially known as DowFroth 250® (usually abbreviated as DF250 or similar). Although less commonly used, other frothers such as methyl isobutyl carbinol (4-Methyl-2-pentanol) or MIBC (alcohol frother) and cresylic acid (phenol-type frother) have also been reported for this application (Table 2). It should be noted that both pine oil and cresylic acid have gradually fallen into disuse in the minerals industry in general, the latter being more environmentally concerning (Khoshdast and Sam, 2011, Fuerstenau et al., 2007). On the other hand, MIBC has become more popular in recent decades despite its potential health risks and strong odour (Fuerstenau et al., 2007). Most of the aforementioned frothers perform well in both acidic and alkaline environments; notably, cresylic acid only has optimal frothing properties in the acidic pH spectrum (Bulatovic, 2007, Khoshdast and Sam, 2011). Frother dosage, in general, has typically been lower relative to that of other flotation reagents, usually ranging from 10 to 100 g/t. In some instances, staged frother addition has been required, with heavier dosage in the rougher stage (Table 2).
Table 2. List of frothers reported in stibnite flotation literature. 1.