1. Introduction
More than four thousand years ago, Egyptian physicians started using drugs in the form of pills, ointments and salves as treatments for illnesses. The first intravenous injection was done in human in 1665 and subcutaneous injections were introduced in 1853 [1]. Subsequently, the modern hypodermic syringe was developed in 1884. The current means of drug administration still have close resemblance with these ancient methods and undergo little change throughout the years. Some of these administration means developed over the years have specific advantages for particular agents or certain diseases [2]. Table 1 shows a brief summary of the common routes of drug administration [3].
Table 1. The common routes of drug administration.
| Administration routes | Examples | Pros | Cons |
|---|---|---|---|
| Intravenous injection | Antibiotics for sepsis | 100% bioavailability | Discomfort to patient, Requires health care provider, Risk of overdose or toxicity, Risk of infection |
| Intravenous infusion | Heparin for anticoagulation | 100% bioavailability, Continuous control over plasma levels | Requires hospitalization, Risk of infection |
| Subcutaneous injection | Usually high bioavailability | Discomfort to patient | |
| Intramuscular injection | Insulin for diabetes | Usually high bioavailability | Discomfort to patient |
| Oral | Aspirin, acetaminophen, ibuprofen | Convenient, Self-administered | Drug degradation prior to absorption, Limited absorption of many drugs |
| Sublingual or buccal | Nitroglycerin for angina | Avoids first-pass metabolism in liver, Self-administered | Limited to lipophilic highly potent agents |
| Ophthalmic | Pilocarpine for glaucoma | Local delivery, Self-administered | Discomfort to some patients, Frequent administration |
| Topical | Antibiotic ointments | Local delivery, Self-administered | Limited to agents that are locally active |
| Intra-arterial injection | Chemotherapy, in some cases | Control of vascular delivery to specific regions | High risk |
| Intrathecal injection | Pain medication, in some cases | Direct delivery to brain | Limited drug penetration into brain tissue, High risk |
| Rectal | Avoids first-pass metabolism in liver, Self-administered | Discomfort leads to poor compliance in some patients | |
| Transdermal | Nitroglycerin patches for angina | Continuous, constant delivery, Self-administered | Skin irritation, Limited to lipophilic, highly potent agents |
| Vaginal | Spermicides | Self-administered | Discomfort leads to poor compliance in some patients |
| Controlled release of implants | Norplant for contraception | Long-term release | Requires surgical procedure |
(Reproduced from ref. [3] with permission.)
Research and developments in creating new and innovative drug delivery systems are increasing at a fast pace globally due to the increasing demands in low cost and higher efficiency for better therapies [4]. To meet this demand, many well-known and effective applied drugs will be reformulated in new drug delivery system to provide enhanced efficiency or more beneficial therapy. One of the most promising drug delivery systems is nanotherapeutic delivery system [5]. It is a drug delivery concept in nanoscience and it is often refers to as an strategy that develops platforms and nanoscales devices for selective delivery of therapeutic genes as well as small drug molecules to the cells of interest [6]. In this paper, the release mechanisms of the drug in nanotherapeutic delivery systems will be covered based on the linker types for the various drugs that are conjugated to nanocarriers. A discussion on how the mechanisms are designed and used in the delivery platform to provide specific targeted release to specified cells will also be mentioned.
1.1. Nanocarriers based therapeutic delivery
There are infinite possibilities to the application of nanotechnology in current drug delivery systems. Most biological functions are highly dependent on nanoscale dimension units like viruses, ribosomes and molecular motors [7]. Thus by having nanoparticles that are small enough for direct interaction with subcellular compartments, it opens up the possibility of activating intracellular events [8].
In general, the nanocarriers based therapeutic drug delivery systems are preferred over normal drug delivery due to the numeral promising advantages [9]. Firstly, the larger surface-to-volume ratio of the nanocarriers could allow a larger contact area of the drug with the body in the same drug concentration condition, thus the same dose of drugs makes the delivery more effective [10]. The reduced dosage will in turn reduce the drug's side effects and toxicity issues. Secondly, nanocarriers have tuneable surface chemistry for different drugs and targeting molecules. For example, the highly efficient drug doxorubicin (DOX), which is used to treat various types of tumours also has a major side effect of causing serious cardiotoxicity due to the lack of tumour specific cytotoxicity. The tuneable surface chemistry allows the possibility of having prolonged and sustained drug release, this improves the bioavailability of the drug to where and when it is most needed and also allows longer period of drug circulation than the drug alone [7]. For instance, one can make drugs that are hydrophobic and has low aqueous solubility to be transported in vivoconditions. The surface chemistry modification allows efficient navigation in the complex in vivo environment by protecting the drug from undue degradation. The surface chemistry modification also enables not only the drug to be directed to specific cell types for targeted delivery but even to special regions of the cell making enhanced intracellular trafficking of the drug possible [11]. Lastly, the nanocarriers can provide the flexibility in the forms of having more diverse routes of drug administration and also in terms of drug formulation.
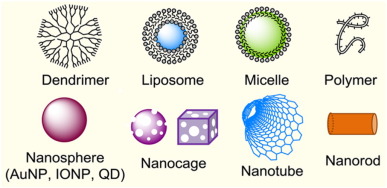
The nanocarriers that carry the drugs can be made in different forms like dendrimer, liposome, rods and many other forms. These nanocarriers can be organic based dendrimer nanoparticles (NPs) [12], polymer [13], polymer based micelles [14], hydrogels [15], [16] or inorganic based like gold nanoparticles(AuNPs), magnetic nanoparticles [17], semiconductor nanoparticles [18], carbon nanotubes(CNTs) or even ceramics like iron oxidenanoparticles (IONPs) [19]. The different forms of nanocarriers are as shown in Fig. 1 [20]. They are versatile and often have many different functions. Normally the therapeutic agents are dissolved, adsorbed, entrapped, encapsulated or attached on the surface or inside the nanocarriers. These nanocarriers are currently used as targeted nanotherapeutics and also in diagnostic test for inflammatory, infectious and autoimmune diseases as well as cancer [21], [22], [23]. When the nanocarriers are in the blood streams, the plasma proteins, cells and other blood components will interact extensively with the material, some of the interactions aids the transportation of the nanocarriers with the drugs to the targeted sites [24].

Fig. 1. Schematic illustration showing the different types of nanocarriers used in drug delivery.
(Reproduced from ref. [20] with permission.)The nanocarriers can be modified to have its own chemical and physical properties by altering the synthesis method, surface functionality and modification, size, shape and the bulk structure [25]. To develop an effective therapeutic delivery system, one needs to know the physicochemical properties of the material and how it interacts with the biological systems in our body [26]. For instance, optimal surface modifications are required to eliminate or reduce undesirable effects like intrinsic toxicity and immunogenicity, which some of the nanomaterials may have. Using cationic nanoparticles as an example, they are cytotoxic due to its nature to disrupt cellular membranes, but by modifying the surface with neutral or anionic groups we can reduce or eliminate the cytotoxicity [13], [27]. By doing surface modification, many of the inorganic materials like cobalt-chromium nanoparticles which are originally toxic in the body can be used. Similarly, the properties of the nanocarriers are custom made for the effective drug loading and efficient mechanisms for release of the therapeutic agent [28].
The main function of these nanocarriers is to transport the therapeutic drugs to the targeted site. The process involved in the cellular delivery of therapeutic agents usually involved passive diffusion, particle phagocytosis, pinocytosis or receptor-mediated endocytosis [3]. This is done by either passive targeting approach or active targeting approach [29]. Fig. 2 shows the schematic illustration of drug carrying nanoparticles in tumour directed delivery based on the active and passive targeting. Passive targeting makes use of increase in endothelial blood microvasculature permeability in tumours due to the larger interstitial gaps between the neighbouring cells to facilitate the delivery of the drug loaded nanocarriers into the tumours [30]. This enhanced permeation and retention (EPR) effect allows greater accumulation of drugs as well as longer drug exposure duration in the tumour due to limited elimination [31]. Active targeting approaches make use of EPR effect and the targeting ligands that are covalently bonded to the nanocarriers surface to target on specific cancer cells. These targeting ligands are specific for a particular cell surface biomarker or over-expressed receptor molecules in the cancerous cells [20], [30].

Fig. 2. Schematic illustration of drug carrying nanocarriers in (A) passive targeting and (B) active targeting in tumour-directed delivery. Ligands attached to the surface of nanocarriers bind to receptors over/expressed by cancer cells or endothelial cells.
(Reproduced from ref. [29] with permission.)The common surface biomarkers that are overexpressed in cancers and inflammatory diseases can be classified under vitamin receptors, αvβ3 integrin receptor, PSMA (prostate-specific membrane antigen) receptor, growth factor receptor, insulin and insulin-like receptors, selecting protein molecules and transferrin [20]. Under the vitamin receptors family are folic acid (FA, vitamin B9) receptors (Far-α, FAR-β), riboflavin (vitamin B2) receptor and biotin receptor and under the growth factor receptors family are fibroblast growth factor receptor (FGFR), Her2 as well as epidermal growth factor receptor (EGFR). The targeting ligand is usually attached in multiple copies to the nanocarrier surface since the mechanism for endocytic uptake of these targeted carriers requires the combined occurrences of several concurrent interactions at the contact point of many pairs of surface receptor and ligand [32]. As a result of the multivalent binding mechanism, tight adhesion between the nanocarriers and targeted cell surface is obtained during the receptor-mediated uptake of the nanocarriers by the targeted cell [25], [33].
2. Drug release mechanisms through linker cleavage
This paper covers on the nanocarrier systems that use different covalent links to attach and carry the therapeutic molecules as well as those that carry the unmodified drug molecules by encapsulation or complexation. The importance of the linkers that serves as a covalent attachment for carrying the therapeutic molecules as well as how the linkers are used to control the release mechanism of the drug are highlighted. The insights on the drug release mechanisms will be discussed based on the structure and functions of the different types of linkers. In addition, the recently developed mechanisms of using photochemistry or thermolysis to trigger drug release in an actively controlled manner and their applications will also be discussed.
2.1. Cleavage by hydrolysis
2.1.1. Hydrolysis of ester bond
Carboxylic ester based linkers are used in the targeted drug release mainly due to the wide use of ester linker to attached to many therapeutic molecules and convenience. The conjugation or ester based drug attachment is formed between the nanocarrier and drug by having a pair of functional groups like carboxylic acid or alcohol [34]. The ester bond formed allows the drug release to take place due to its susceptibility to the hydrolysis process in vivo under physiological conditions. This hydrolytic reaction is catalyzed by the presence of acids, bases, metal ions like copper(II) ions and hydrolytic proteins like esterases and human serum albumin. Owing to the physiological instability of ester bond in vivo, it is not effective and specific enough to allow controlled drug release to take place.
One of the main process of drug delivery is receptor-mediated endocytosis where the nanocarriers are engulfed into the cytosol through coated pits and become part of the endosome before it is being fused with lysosomes [35]. During the process of endocytosis, numerous acid hydrolases like cholesteryl ester acid hydrolases, aryl sulfatase, acid phosphatase, N-acetylglucosaminidase and cathepsin D are present [36]. These hydrolases will aid the ester or amide hydrolysis including ester based linkers between the drug and nanocarriers. More optimal catalytic activity will take place causing drug release to occur more rapidly and selectively after uptake if the environment caused by the endosomes is between pH 5.0 to 6.0 and lysosomes is at pH 4.8 instead of the neutral environments like plasma and cytoplasm with pH 7.4 [37]. In short, the mechanism of cleaving of ester linkers that causes drug release is strongly connected to the specific uptake by the targeted cell and the intracellular linker hydrolysis which is catalyzed by the acid hydrolases in the endosomes and lysosomes [36]. There are other hydrolytic enzymes that associated with the disease and overexpressed in the plasma or extracellular membrane. Due to the abnormalities in diseased cells under pathological conditions, it can cause upregulation in expression producing enzymes that catalyse the hydrolysis of the ester linkers causing drug release to take place. For instance, a Zn2 +/Mg2 +dependent membrane-bound metalloenzyme, alkaline phosphatase is proven to release a quinolinone drug molecule by acting as catalyst for the hydrolysis of the drug to O-phosphate bond [38]. In addition, a serine-dependent esterase, Carboxylesterase 2 (CE-2) found on the membrane comprises one of the biomarkers proteins that is expressed in abundances in liver and colon tumour. It will catalyse the hydrolysis of endogenous lipid esters and also cleavage of carbamate-based prodrug substrates like irinotecan (CPT- 11), an anticancer agent causing drug release [39]. The cleavage of CPT- 11 releases SN38 as an active metabolite which inhibits topoisomerase I [39].
On the other hand, the chemical mechanism like hydrolysis reaction will also cause the ester linkers to be cleaved and is catalyzed by the presence of acid, base and metal ions as mentioned previously [40], [41]. Most esters are actually more stable in slightly acidic condition of pH 5 to 6 as ester hydrolysis through an acid or basic catalyst has the least impact at this pH range. Thus the drug release caused by chemical hydrolysis of the ester linkers that bonds the drug to nanocarriers is insignificant even after the uptake into acidic endosomes and lysosomes or on exposure to the extracellular matrix (ECM) of the tumour which has a pH of 6.2 to 6.9. This ester linker has high susceptible to base-catalyzed hydrolysis which allows controlled drug release to take place in basic subcellular compartments like mitochondrial matrixes which have a pH of 7.9 to 8.0 and peroxisomes which has a pH of 8.2 [42]. It was observed that the rate of hydrolysis of ester linkers in the ester conjugates of paclitaxel is 10 times faster with every one unit increase in pH [43].
Control of drug release through ester hydrolysis mechanism has been widely exploited in previous investigations, including Taxol, Methitreate, Platinum agents and SN38. For example, the taxane class of anticancer agents like paclitaxel and docetaxel are classified as microtubule-stabilizing agents since they are able to bind and stabilize cytosolic microtubules [44]. The microtubules are hollow cylindrical polymers which are made up of repeating units of heterodimer, α-tubulin and β-tubulin [45]. On the luminal side of the tubular structure, the taxol molecule will bind selectively to the β-tubulin unit. This stabilization of cytosolic microtubules inhibits cell growth by impeding with the cellular division process and the mobility which is highly dependent on the microtubules' assembling and disassembling ability. An ideal taxol drug delivery system requires a mechanism of drug release after the drug conjugate is uptaken by the cells [46]. The taxol are conjugated to the nanocarriers by using the secondary alcohol at the C2 position or linked by ester bond at C10 position. An example of a clinically certified taxol conjugate is polyglumex (Xyotax) which is made of biocompatible poly(L-Glu) polymer. Each of the polymer molecules consists of a few paclitaxel molecules which are linked by an ester linkage at the C2 position to γ-carboxylic acid in the polymer [47]. The drug will travel into the cancer cell through endocytosis and gets released by lysosomal enzymes like cathepsin B. However, it was also reported that is too unstable as can be degraded rapidly and release the drug even without enzymes. Other than direct chemical hydrolysis and enzymatic hydrolysis of the ester linkage, the release of taxol can take place by ester hydrolysis through the addition of disulfide group next to an ester linkage [48], [49]. Other types of chemicals used are single walled carbon nanotube (SWNT) and triazine dendrimer. For instance, Dai et al. demonstrated the conjugation of PTX to branched poly(ethylene glycol) (PEG) chains on SWNTs via a cleavable ester bond to obtain a water soluble SWNT-paclitaxel conjugate (SWNT-PTX) (Fig. 3) [48]. The authors noted that the SWNT-PTX formulation afforded higher efficacy in suppressing tumour growth than clinical Taxol® in a murine 4T1 breast-cancer model. This is because of the prolonged blood circulation and 10-fold higher tumour PTX uptake by SWNT delivery. It showed that the drug molecules carried into the reticuloendothelial system could be released from SWNTs and further excreted via biliary pathway without causing obvious toxic effects to normal organs, indicating promising potential for high treatment efficacy and minimum side effects for cancer therapy [48].

Fig. 3. Schematic illustration of paclitaxel conjugation to SWNT functionalized by phospholipids with branched-PEG chains. The PTX molecules are reacted with succinic anhydride (at the circled OH site) to form cleavable ester bonds and linked to the termini of branched PEG. This allows for releasing of PTX from nanotubes by ester cleavage in vivo.
(Reproduced from ref. [48] with permission.)Methotrexate (MTX) is an important drug under the antifolate molecules family which are currently used to treat cancers and inflammatory diseases. When used in subnanomolar concentration, it shows signs of inhibition of cytosolic dihydrofolate reductase [50]. This dihydrofolate reductase acts as a catalyst in the reduction of dihydrofolate to tetrahydrofolate. The tetrahydrofolate is a cofactor involved in the de novo biosynthesis of thymidine and also linked to the building block in DNA. However, MTX has a narrow therapeutic index owing to its dose-limiting systemic toxicity, hence it is more suited for usage in targeted drug delivery system [51]. There are two carboxylic acids present on the L-Glu portion of each MTX molecule, they are both able to be used to covalently bond nanocarriers by ester linkages. Recently, Majoros et al. reported that the possibility of conjugating MTX to PAMAM dendrimer fifth generation (G5) by EDC based ester linker. This fifth generation PAMAM dendrimer was also conjugated to folic acid receptor targeting ligand, FA. In vitro, the uptake in conjugate through FAR- positive KB cell line undergo receptor-mediated endocytosis shows possibility of inhibiting cell multiplication (Fig. 4) [52]. In vivo, the conjugate demonstrated improvement in therapeutic efficacy in treatments for epithelial cancer, head, neck tumours and inflammatory arthritis. However, the exact mechanisms of action and release for MTX conjugated by ester linkage is yet to be known. It is important to highlight that in acidic conditions that is similar to the intracellular environment of endosomes, the ester conjugate of MTX displayed great resistance against the hydrolysis process which may affect the cellular uptake. Other MTX delivery systems that make use of ester linkages are glycidylated PAMAM dendrimer which are FA conjugated, MTX conjugated to G3 PAMAM dendrimer, MTX prepared by “one pot” reaction, MTX conjugated to G5 PAMAM dendrimer by a triazine spacer through azide-alkyne click chemistry [53], [54].

Fig. 4. The diagram showing the conjugation of MTX to G5 PAMAM dendrimerby ester linkage and its anti-tumour mechanisms.
(Reproduced from ref. [52] with permission.)In another aspect, platinum agents are among the most powerful and widely used chemotherapy drugs against cancer. The first chemical of the platinum-based anticancer therapeutic agent family discovered is cisplantin, Pt(II) (NH3)2Cl2. The two amine ligands in cisplantin are strongly chelated to the platinum ion and thus they have low reactivity towards carboxylic acid to form amide linkage [55]. Cisplantin conjugation is done by replacement of the chloride ligand with a linker residue since the two chloride ligands are displaced with water via ligand exchange reaction when placed in an aqueous medium. An example is the conjugation of platinum(IV) agents to a SWNT by carboxylate chelation method [56]. The uptake by the cancer cells takes place due to endocytosis process where pH is less than seven inside the endosomes and lysosomes. This drop in pH enhances the reductive release of the platinum (II) core complex. Due to the high cytotoxicity of SWNT-Pt to cancer cells, the activity increases by 100-folds compared to platinum free drug. This performance is due to the more efficient endocytic uptake and enhanced activation by the drug attached to the SWNT nanocarrier. Different from platinum-based chemotherapy drugs, SN38 is an active product in the metabolism of irinotecan which has shown great potential as anti-tumour drug in targeted drug delivery due to its insolubility in water. PAMAM terminated with carboxylic acid group was also used as a carrier for SN38 by attaching with ester linkers and glycine or β-alanine spacer at the C20-OH position [57]. The small difference in structure by a CH3 group between glycine and β-alanine spacer affects the conjugates activity as well as the rate of drug release. The release of SN38 with glycine is higher than that with the β-alanine causing SN38 with glycine to be more potent. Recently, Gu et al. reported the controlled release of SN38 via thiolysis in the presence of GSH (glutathione) or viaenhanced hydrolysis due to ROS (reactive oxygen species)-oxidation of the linker, giving rise to high in vitro cytotoxicity and in vivo anticancer therapeutic activity (Fig. 5) [58]. By tuning the length of the OEG chain in the prodrug, the obtained amphiphilic conjugates were capable of self-assembling into nanocapsules. In vivo studies showed the conjugated SN38 had higher tolerance than the free drug and gave rise to higher in vitro cytotoxicity and in vivoanticancer therapeutic activity. Immunosuppressive agents like tacrolimus (FK506) and cyclosporine are important therapeutic drug in organ transplantation. FK506 is more potent than cyclopsporine by two orders under in vitro conditions, however it is also rapidly cleared in plasma after intravenous administration and highly metabolized by P450 3A liver cytochrome enzyme. Thus to control the pharmacokinetics and enhance the therapeutic efficiency, Hashida et al. developed a delivery system using dextran to transport FK506 by conjugating with ester linker. Owing to the fairly stable ester bond, the rate of drug release became lower in a phosphate buffer of pH 7.4 with a half-life of around 150 h and an extended circulation time in the bloodstream [59]. Abdi et al. created a targeted delivery system to carry cyclosporine by coprecipitating cyclosporine-tethered polylactide with PEG linked polylactide [60]. The nanoparticles are internalised in the dendritic cells and move to lymph nodes where the cyclosporine is released in a sustained manner when ester linkage is broken causing the T-cell proliferation to be suppressed [60].

Fig. 5. Structure of GSH and the ROS-responsive conjugate of SN38. The conjugate self-assembled as nanocapsules that can release SN38 with higher in vitro cytotoxicity and anticancer therapeutic activity.
(Reproduced from ref. [58] with permission.)2.1.2. Hydrolysis of amide bond
In addition to the hydrolysis of ester bonds, amide linkers are also commonly used as a conjugation between the drug and nanocarriers due to the ease of functionalising the drug molecules with an amine or carboxylic acid group and the stability of amide bonds to chemical hydrolysis compared to ester bond [61]. However, this stability causes the amide bonds to be rarely chemically hydrolyzed under physiological conditions, instead harsh conditions like higher temperatures and the presences of strong acid or base catalysts is required. This gives it a better pharmacokinetic profile due to the longer circulation duration in the bloodstream [62].
Generally, most amide cleavage is done using enzymatic mechanisms by hydrolytic proteases like serine proteases, cysteine proteases and zinc-dependent endopeptidases [63]. These enzymes are found in various sites like extracellular environment and cell membrane. The site of drug release is closely linked to the site of specific action. For instance, the matrix metalloproteinases (MMPs) such as collagenases are secreted by zinc endopepridases in extracellular matrix (ECM) due to degradation of proteins [64]. In some of the tumour cells, the MMPs are overexpressed and implicated in the dysregulation of angiogenesis causing the growth and spreading of tumour cells [65]. By using a MMP-specific peptide as the cleavage bond, controlled drug release can be obtained at ECM tumour site. In addition, prostate-specific antigen, PSA, is from the serine protease family. It is expressed at high levels in prostate tumour tissue and also used in controlled drug release as an enzyme target. The sequences in the peptide consisting serine (S)-glutamine (Q) and glutamine (Q)-leucine (L) are most susceptible to PSA [66]. The peptide spacer comprising SQ dipeptide sequence in aminoglycoside is conjugated to doxorubicin which is readily released by PSA through peptide cleavage [66]. Prostate-specific membrane antigen which is a zinc endopeptidase, is another enzyme biomarker that is expressed in large amount in prostate cancer cells [67]. On small molecule substrates like folate-γ-polyglutamate (FA-En), methotrexate-γ-polyglutamate (MTX-En) and neuropeptide N-acetyl-L-aspartyl-L-glutamate (AC-DE), the PSMA can act as a glutamate carboxypeptidase (GCPII) by having it in the linkage will allow possibility of targeted drug release in prostate tumours. Another protease that can be part of the peptide linkage conjugated to the drug is plasmin [68]. Plasmin similar to PSA is a serine protease which is found in plasma and lysosomes.
On the contrary, there is no general mechanism for drug release through cleavage of amide linkage by chemical hydrolysis but some classes of specialised linkages conjugated to the drug by amide linkage are responsive to low pH environments [69]. The specialised linkage contains a form of maleic acid like citraconyl, cis-aconityl and maleyl group, it is cleaved in an acid-catalyzed mechanism to release the drugs. At pH 7, this linkage is very stable and does not hydrolysed much even after four days of incubation. However, the rate of hydrolysis will increase with increasing acidity, this allows controlled realise of the drug by varying the pH of the environment. Previous study has shown that the cis-aconityl linker hydrolysed significantly faster than the maleyl linker in the same environment while citraconyl linker are slightly faster than cis-aconityl linker [70].
Several anticancer drugs such as MTX, DOX and α-Tocopheryl succinate have been conjugated onto different delivery carriers and controlled drug release profiles were achieved by the hydrolysis of the amide bond. For example, methotrexate (MTX) has been used in several cancer targeted treatment through conjugating with a peptide based amide linkage. The drug conjugation is done by having an amide spacer that is peptidase susceptible. The drug release is controlled by the mechanism that depends on the overexpressed peptidases on the cancer cell surface. Recently, a dextran-based delivery system is designed by Langer et al. to deliver MTX that is peptide linked and susceptible to matrix metalloproteinases (MMPs) [71]. The efficiency of drug release is sensitive to the type of peptide space as well as MMP subtype. A lower level of release (61%) in 24 h is observed for treatment done using MMP 9 and MTX is not released when it is directly conjugated to dextran or by random peptide sequence [71]. Similar to dextran, the MMP specific peptide spacer is used in FAR-targeted MTX delivery through PEG-poly(lysine)-grafted dendrimers. The peptide linked MTX is also used with MWNT and hyaluronic acid (HA). Bianco et al. used MWNT that was attached to MTX via amide-based spacer with varied peptide length and sequence and the uptake by the breast cancer cells was based on folate receptor-mediated endocytosis. Results showed that only specific conjugates that are linked by GFLG peptide sequence could inhibit cell proliferation [72]. In another example, Homma et al. used a small set of short peptides for conjugation between MTX and HA backbone for the treatment of osteoarthritis as MTX is a general cytotoxic chemical for cancer and rheumatoid arthritis treatment. The results also showed the need for specific peptide for MTX release [73].
Doxorubicin is conjugated by amide or peptide based linkers that are made by attaching daunosamine sugar residue so that it is cleaved by peptidase action or low pH. Jones et al. used peptide containing LSQ sequence that is hydrolysed between S and Q site by prostate-specific antigen (PSA) to release doxorubicin [74]. When expose to prostate tumour cells that secrete PSA, the drug release will produce free doxorubicin and doxorubicin-leucine. This allows tumour selective drug delivery as the doxorubicin is more cytotoxic to cancer cells secreting PSA than those that do not.
In a recent study, Zhang et al. demonstrated the fabrication of DOX-Fe3O4conjugates with the ability to release the conjugated drug by cleavage of amide bond in weakly acidic environment (Fig. 6) [75]. The DOX-Fe3O4 nanoparticles were further embedded in a polyethylene glycol (PEG) functionalized porous silica shell (Fe3O4-DOX/pSiO2-PEG) to achieve the controlled drug release. In addition to the cleavage of chemical bond, the presence of a porous silica shell could provide a protective layer for drug molecules and magnetite nanoparticles. Furthermore, the porous silica shell imposed a further obstacle for the DOX release from the carrier, and thus it exhibited a slower release rate than the uncoated DOX-conjugated Fe3O4 nanoparticles. The bioassay showed that when the nanoparticles were transported to early endosomes, the endosomes could fuse with low pH lysosomes to cleave the amide bond between DOX and the particle. This allows the DOX to be released first from the Fe3O4 particle surface and then to enter via the pore channels into the nucleus of the target cell, where it performs antitumor activity [75].
